
Abstract
Spinal muscular atrophy (SMA) is characterized by degeneration of spinal motor neurons and is a leading genetic cause of pediatric death worldwide. SMA results from the loss of or pathological variant in the survival motor neuron 1 (SMN1) gene. Disease severity is dependent on the number of copies of the orthologous SMN2 gene, which is nearly identical to SMN1 except for some key nucleotide differences. As disease severity is inversely related to SMN2 copy number, most SMA therapeutics trials have focused on identifying ways to increase SMN2 expression at different levels of gene regulation. Other studies have investigated compounds which protect affected motor neurons and their target muscles in an SMN-independent manner. In this study, we examined the therapeutic efficacy of the effect of a combination regimen of the SMN2 inducer D156844 and the neuroprotective agent AR42 (REC-2282) on the disease progression and survival in the SMNΔ7 SMA mouse model. The dual administration of D156844 and AR42 results in an additive improvement in the survival of these mice as well as delaying disease endstage. Additionally, coadministration of D156844 and AR42 produced improvements in motor phenotype in SMNΔ7 SMA mice. This study provides further evidence underlying the potential benefit of a combination therapeutics approach to treating SMA.
Similar content being viewed by others


Introduction
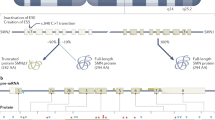
Spinal muscular atrophy (SMA), a leading genetic cause of death in infants and children, is an autosomal recessive degenerative disease characterized by selective loss of α motor neurons of the anterior horn of the spinal cord1. There is a marked atrophy of limb and trunk muscles in SMA patients because of motor neuron loss. SMA results from the loss of or pathological variant in the survival motor neuron (SMN) gene2. The SMN gene is duplicated in humans (SMN1 and SMN2) and SMN1 and SMN2 differ by a single C-to-T transition within an exon splice enhancer of exon 7 3,4. The SMN1 transcripts contain exon 7 to produce full-length SMN protein while most SMN2 transcripts lack exon 7 to yield an unstable protein known as SMNΔ7. The number of copies of SMN1 and SMN2 are variable within the human population. The copy number of SMN2 modifies the severity of SMA phenotype in humans5,6,7 as well as in transgenic mouse models for SMA8,9,10 demonstrating that SMN2 is a genetic modifier of disease phenotype.
There are currently three different FDA-approved SMN-inducing therapies for SMA: nusinersen (Spinraza)11,12,13risdiplam (Evrysdi)14 and onasemnogene abeparvovec (Zolgensma)15. Current treatments for SMA are effective in terms of increasing lifespan and improving motor function but there still remains significant disability as well as variable responsiveness, uncertain long-term efficacy20 and high economic burden21,22. As such, finding small molecule drugs that can increase SMN2 expression or support motor function in an SMN-independent manner provides an opportunity to maximize therapeutic benefits of these current treatment options.
Many studies have identified several chemically distinct classes of compounds that increase SMN2 expression23. C5-substituted 2,4-diaminoquinazolines (2,4-DAQs) are potent inducers of SMN2 promoter activity that were initially identified through a high-throughput drug screen24. D156844, a piperidine 2,4-DAQ derivative, that is orally bioavailable and increases SMN2 expression in cultured fibroblasts derived from an SMA patient and ameliorates the survival and phenotype of SMNΔ7 SMA mice25,26,27. RG3039, another potent 2,4-DAQ, increases the mean lifespan in different mouse models of SMA28,29. The 2,4-DAQs tested in vivo in SMA mouse models, however, did not have marked effects on SMN2 expression suggesting that either the effect of these agents on SMN2 expression was below the detection threshold in mice or these agents exert their protective effects on SMA mice independently from regulating SMN2 expression.
Histone deacetylase inhibitors like trichostatin A (TSA) and 4-phenylbutyrate (4-PBA) have been shown to increase SMN2 expression in cell culture model for SMA30,31,32. When TSA or 4-PBA were administered pre-symptomatically, there were significantly improvements in the survival and motor phenotype of SMA mouse model30,33,34. It is unclear whether these compounds affect SMN2 expression in vivo. TSA is not orally bioavailable. Additionally, TSA and 4-PBA require fairly high dosing concentrations to exert their protective effects in mice. These limitations of TSA and 4-PBA lead to the discovery of potent analogues. AR42 (OSU42; REC-2282) is a TSA:4-PBA hybrid molecule that is orally bioavailable and has potent HDAC inhibitor activity35,36. AR42 significantly improves the survival and motor phenotype of SMNΔ7 SMA mice when administered pre-symptomatically but does not increase SMN2 expression in vivo even though AR42 increases SMN2 expression in SMA fibroblasts37.
A combinatorial approach has been suggested as a plausible option to maximize therapeutic benefit in SMA38. This approach would administer different classes of therapeutic agents to elicit a multi-faceted ameliorative effect on SMA patients. Ideally, this combinatorial approach would take the form of a cocktail of therapeutic agents that target multiple mechanisms underlying SMA pathology. Combination therapeutics strategies based on targeting two different aspects of SMN gene expression39,40targeting SMN expression and a SMA modifier gene like neurocalcin D (Ncald)41 as well as targeting SMN expression and neuroprotection or myoprotection42,43,44 have demonstrated additive or, in some cases, supra-additive therapeutic efficacy in SMA mouse models. Even assessing the effect of diet on therapeutic responsiveness27,33 can be viewed as a combination therapeutic strategy. In this study, we examined the therapeutic efficacy, in terms of survival and motor function, of a combination of an orally bioavailable SMN inducer (D156844) and an orally bioavailable neuroprotectant (AR42) in SMNΔ7 SMA mice. This study sets the stage for a detailed analysis of the mechanisms of therapeutic success—beyond SMN2 expression—thereby potentially opening the door for refinement of current strategies.
Results
Effect of diet on survival of SMNΔ7 SMA mice with AR42
As diet affects the responsiveness of SMNΔ7 SMA mice to therapeutic agents liked D156844 27, we determined the effect of diet on the responsiveness of SMNΔ7 SMA mice to AR42. SMNΔ7 SMA mice reared on PicoLab20 Mouse diet showed a significant increase (19.4%) in mean lifespan when compared against vehicle-treated SMNΔ7 SMA mice reared on the same diet (Fig. 1; 16.6 ± 0.7 d vs. 13.9 ± 0.6 d; χ2 = 6.535, p = 0.011). The therapeutic benefit of AR42 on SMNΔ7 SMA mice reared on the PicoLab20 Mouse diet was similar to that of AR42-treated SMNΔ7 SMA mice reared on the Harlan-Teklad diet (previously published37). Comparison with previously published, AR42/Harlan-Teklad data is valid since both groups originated from the same mouse colony, were maintained on the same diet and received the same environmental conditions. Furthermore, the mean lifespan of vehicle-treated SMNΔ7 SMA mice in this study was not different from the previous work using this mouse colony (13.9 ± 0.6 vs. 11.4 ± 0.4 d)27,42.

Oral administration of AR42 increased the survival of SMNΔ7 SMA mice that were maintained on the PicoLab20 diet. Kaplan-Meier survival plot for SMNΔ7 SMA mice receiving either vehicle on a PicoLab20 diet (dotted blue line; n = 17), 10 mg/kg/d AR42 on a PicoLab20 diet (dashed orange line; n = 16), vehicle on a Harlan-Teklad diet (dotted-dashed red line; n = 15) or 10 mg/kg.d AR42 on a Harlan-Teklad diet (solid green line; n = 15). Treatment of SMNΔ7 SMA mice began at PND04. The data for the mice maintained on the Harlan-Teklad diet were previously published37.
Effect of AR42 and D156844 coadministration on the survival of SMNΔ7 SMA mice
As oral administration of either D156844 27 or AR42 37 significantly increased the lifespans of SMNΔ7 SMA mice, we determined the effect of coadministration of D156844 and AR42 (D156844 + AR42) on the survival of SMNΔ7 SMA mice. The dosing of these mice began at PND04. We did not observe any issues with safety or tolerability in neonatal mice treated with the combination of D156844 and AR42. Coadministration of AR42 and D156844 resulted in a 71% increase in mean lifespan relative to vehicle-treated SMNΔ7 SMA mice (Fig. 2; Table 1; 23.7 ± 3.5 d for D156844 + AR42 combo (brown solid line) vs. 13.9 ± 0.6 d for vehicle (blue dotted line); χ2 = 16.986, p < 0.001). The ameliorative effects of D156844 + AR42 on the survival of SMNΔ7 SMA mice were greater than either D156844 27 (Fig. 2; Table 1; 23.7 ± 3.5 d for D156844 + AR42 combo vs. 17.7 ± 0.6 d for D156844 (gray solid line); χ2 = 4.856, p = 0.028) or AR42 alone (Fig. 2; Table 1; 23.7 ± 3.5 d for D156844 + AR42 combo vs. 16.6 ± 0.7 d for AR42 (blue dotted line); χ2 = 6.771, p = 0.009).

Oral administration of AR42 augmented the D156844-induced increase in survival of SMNΔ7 SMA mice. Kaplan-Meier survival plot for SMNΔ7 SMA mice receiving either vehicle (dotted blue line; n = 17), 3 mg/kg/d D156844 (solid gray line; n = 13), 10 mg/kg/d AR42 (dashed orange line; n = 16) or a combination of 10 mg/kg/d AR42 and 3 mg/kg/d D156844 (solid brown line; n = 15). Treatment of SMNΔ7 SMA mice began at PND04. All mice within the treatment cohort were maintained on the PicoLab20 diet.
Effect of D156844 and AR42 on disease progression in SMNΔ7 SMA mice
The onset of body mass loss is an indicator of the final stages of disease in the SMNΔ7 SMA mice45,46. Cotreatment of SMNΔ7 SMA mice with D156844 + AR42 significantly delayed the onset of body mass loss by 63%, relative to vehicle-treated SMNΔ7 SMA mice (compare brown solid line vs. blue dotted, line in Fig. 3; 18.6 ± 3.3 d vs. 11.4 ± 0.4 d; χ2 = 18.222, p < 0.001, Table 2). AR42 treatment (orange dashed line in Fig. 3) alone also significantly delayed the onset of body mass loss in SMNΔ7 SMA mice (13.0 ± 0.5 d vs. 11.4 ± 0.4 d; χ2 = 5.213, p = 0.022). The D156844 + AR42-induced delay in onset of body mass loss in SMNΔ7 SMA mice was greater than that for either D156844 27 (Fig. 3; Table 2; 18.6 ± 3.3 d for D156844 + AR42 vs. 12.5 ± 0.4 d for D156844 (gray solid line); χ2 = 10.986, p < 0.001) or AR42 alone (Fig. 3; Table 2; blue dotted line; χ2 = 7.250, p = 0.007).

Oral administration of AR42 augmented the D156844-induced increase in the onset of loss in body mass of SMNΔ7 SMA mice. Kaplan-Meier onset of body mass loss plot for SMNΔ7 SMA mice receiving either vehicle (dotted blue line; n = 17), 3 mg/kg/d D156844 (solid gray line; n = 13), 10 mg/kg/d AR42 (dashed orange line; n = 16) or a combination of 10 mg/kg/d AR42 and 3 mg/kg/d D156844 (solid brown line; n = 15). Treatment of SMNΔ7 SMA mice began at PND04.
The growth curves of SMNΔ7 SMA mice treated with either D156844 or AR42 alone were similar to vehicle-treated SMNΔ7 SMA mice (Fig. 4A; comparing the open diamond, gray curve (D156844) or the closed diamond, orange curve (AR42) against the open squared blue curve (SMA vehicle)). These observations can also be seen upon magnification of these curves during preweaning development (Fig. 4B). In D156844 + AR42-treated SMNΔ7 SMA mice, the growth curve (brown open triangle curve in Fig. 4A and B) was greater than in those SMNΔ7 SMA mice treated with D156844 or AR42 alone as well as with vehicle, especially after PND12. The growth curves of D156844 + AR42-treated SMNΔ7 SMA mice, however, were lower than those of non-SMA littermates (black solid dotted line). Using generalized estimating equations (GEE) analysis47the growth curves of SMNΔ7 SMA mice, regardless of treatment group, were significantly different than those of nonSMA mice (Table 3). Interestingly, the growth curves of SMNΔ7 SMA mice treated with D156844 + AR42 were significantly different from those of SMNΔ7 SMA mice treated with either D156844 alone, AR42 alone or vehicle (Table 3).

The effect of AR42 and D156844 cotreatment on changes in body mass of SMNΔ7 SMA mice. (A) Body mass curves of SMNΔ7 SMA mice receiving 10 mg/kg/d AR42 (orange diamonds), 3 mg/kg/d D156844 (gray open diamonds), 3 mg/kg/d D156844 and 10 mg/kg/d AR42 (brown open triangles) or vehicle (blue open squares) as well as non-SMA littermate (black dots). Treatment began at PND04. (B) Magnification of the growth curves in (A) to better resolve the growth curves for each treatment group during the preweaning phase of the mice. (C) Changes in body mass between PND04 and PND14 of SMNΔ7 SMA mice treated with either 3 mg/kg/d D15644 (gray open diamonds), 10 mg/kg/d AR42 (orange filled diamonds), 3 mg/kg/d D156844 and 10 mg/kg/d AR42 (brown open triangles), or vehicle (blue open squares). The change in body mass between PND04 and PND14 of age-matched non-SMA littermates (black dots) was also shown. The statistically significant differences between pairs of experimental groups were provided above the bar graph.
The growth rate—as measured by the change in body mass between PND04 and PND14—was significantly diminished in SMNΔ7 SMA mice relative to non-SMA littermates, irrespective of treatment group (Fig. 4C;45,46). Interestingly, the growth rate of SMNΔ7 SMA mice treated with D156844 + AR42 was significantly greater than that of AR42-treated SMNΔ7 SMA mice (p = 0.017; compare the brown bar against the orange bar in Fig. 4C). The growth rates between SMNΔ7 SMA mice treated with D156844 + AR42 (brown bar) and those receiving vehicle (blue bar) were not significantly different.
Effect of D156844 and AR42 on the motor phenotype of SMNΔ7 SMA mice
There is a progressive impairment of motor behavior in neonatal SMNΔ7 SMA mice that is characterized by a loss of surface righting reflexes and reduced spontaneous locomotor activity46. As D156844 26 and AR42 37 ameliorate motor impairment of SMNΔ7 SMA mice, we determined the effect of D156844 and AR42 coadministration on motor phenotype. One of the earliest observed phenotypic indicators in SMNΔ7 SMA mice is the inability to right themselves when placed in a prone position (surface righting). In agreement with previous findings46we observed that SMNΔ7 SMA mice had an impairment in the surface righting relative to nonSMA littermates and the surface righting latency was significantly longer for those SMA who could right themselves at PND07 (Fig. 5A) and PND11 (Fig. 5B). At PND07, a greater proportion of the SMNΔ7 SMA mice treated with D156844 (gray solid line in Fig. 5A) or D156844 + AR42 (brown solid line in Fig. 5A) exhibited surface righting responses although they were delayed relative to nonSMA littlermates (black solid line in Fig. 5A). The differences in surface righting success at PND07 between the treatment groups was not statistically significant (Table 4). At PND11 (Fig. 5B), some SMNΔ7 SMA mice exhibited successful surface righting response in all of the treatment groups but the differences between treatment groups were not statistically significant (Table 4).

The effect of cotreatment with AR42 and D156844 on surface righting responses of SMNΔ7 SMA mice. SMNΔ7 SMA mice received either 3 mg/kg/d D156844 (gray solid line), 10 mg/kg/d AR42 (orange dashed line, combination of D156844 and AR42 (brown solid line), or vehicle (blue dotted line) beginning at PND04. Behavior responses were also compared against age-matched non-SMA littermates (black solid line). Kaplan-Meier analysis was completed on surface righting success (time-to-event) at PND07 (A) and PND11 (B). The statistical comparisons from the Kaplan-Meier analyses are provided in Table 4.
A delay in the onset of movement (movement latency) is a motor deficit observed in SMNΔ7 SMA mice. Movement latency was greater in SMNΔ7 SMA mice relative to non-SMA littermates at PND07 (Fig. 6A), PND11 (Fig. 6B) and PND14 (Fig. 6C). SMNΔ7 SMA mice treated with D156844 + AR42 displayed a shorter movement latency than vehicle-treated SMNΔ7 SMA mice, with a more pronounced effect at PND11 and PND14 (compare brown solid lines against blue dotted lines in Fig. 6B and C). Kaplan-Meier analysis (Table 5) revealed that movement latency between D156844 + AR42- and vehicle-treated SMNΔ7 SMA mice were significantly different at PND14 ( χ2 = 6.000; p = 0.014).

The effect of cotreatment with AR42 and D156844 on spontaneous movement responses of SMNΔ7 SMA mice. SMNΔ7 SMA mice received either 3 mg/kg/d D156844 (gray solid line), 10 mg/kg/d AR42 (orange dashed line, combination of D156844 and AR42 (brown solid line), or vehicle (blue dotted line) beginning at PND04. Behavior responses were also compared against age-matched non-SMA littermates (black solid line). Kaplan-Meier analysis was completed on movement success (time-to-event) at PND07 (A), PND11 (B) and PND14 (C). The statistical comparisons from the Kaplan-Meier analyses are provided in Table 5.
The amount of time that the neonatal SMNΔ7 SMA mice were engaged in directional movement (movement duration) was greater in those mice treated with AR42 or D156844 + AR42 than in vehicle-treated SMNΔ7 SMA mice (Fig. 7A). Movement duration of non-SMA mice at PND11 was significant longer than vehicle-treated SMNΔ7 SMA mice. This increase in movement duration was especially pronounced in SMNΔ7 SMA mice at PND11 although differences in the movement durations of drug-treated SMNΔ7 SMA mice were not statistically significant. The movement duration in D156844 + AR42 SMNΔ7 SMA mice at PND14 approached the duration observed in non-SMA littermates but statistical analysis could not be completed at this time point.

The effects of cotreatment with AR42 and D156844 on the motor phenotype of SMNΔ7 SMA mice. SMNΔ7 SMA mice received either 3 mg/kg/d D156844 (gray open diamonds), 10 mg/kg/d AR42 (orange diamonds), combination of D156844 and AR42 (brown open triangles) or vehicle (blue open squares) beginning at PND04. Behavior responses were also compared against age-matched non-SMA littermates (black dots). (A) Duration of movement in treated mice at PND07, PND11 and PND14. There was an arbitrary cutoff time of 61 s. (B) Spontaneous locomotor activity—measured as the number of grids crossed in 1 min—in treated SMNΔ7 SMA mice at PND07, PND11 and PND14. (C) The number of pivots, or 90° turns, made in 1 min in treated SMNΔ7 SMA mice at PND07, PND11 and PND14. Key: *, p ≤ 0.05 when compared against vehicle-treated SMNΔ7 SMA mice.
The net movement—measured by the number of grids crossed within 60 s—of PND07 mice were similar for all genotypes and SMA treatment groups (Fig. 7B). At PND11, SMNΔ7 SMA mice showed less net movement, irrespective of treatment group, than non-SMA littermates. D156844 + AR42-treated SMNΔ7 SMA mice at PND14 displayed greater net movement than vehicle- or AR-treated SMNΔ7 SMA mice. Interestingly, the number of grids crossed in the D156844 + AR42-treated SMNΔ7 SMA mice was similar to non-SMA littermates at this age.
Pivoting behavior was not significantly different between treatment groups as well as between genotypes at PND07 (Fig. 7C). At PND11, pivoting behavior was significantly higher in non-SMA mice than in SMNΔ7 SMA mice, regardless of treatment group. While pivoting behavior at PND14 was reduced in SMNΔ7 SMA mice treated with vehicle or with AR42, relative to non-SMA littermates, the number of pivots in D156844 + AR42-treated SMNΔ7 SMA mice was similar than observed in non-SMA littermates.
Discussion
The three different FDA-approved therapies for SMA (nusinersen11,12,13risdiplam14 and onasemnogene abeparvovec15) have markedly delayed disease progression and improved quality of life for SMA patients. Unfortunately, variable responsiveness within the patient population has been observed and many of these compounds also modulate off-target—with respect to SMN2 expression—transcript levels. For example, risdiplam and its analog branaplam also modulate CAG triplet repeat expansion within the huntingtin gene in cells derived from patients with Huntington’s disease16,17. Furthermore, reproductive complications have been observed in male rats and primates treated with risdiplam48 as well as possible peripheral neuropathy in larger animals49thereby calling into question the safety of these current SMA therapeutics. Additionally, the long-term efficacy of existing therapeutic options, like onasemnogene abeparvovec, is uncertain in more severe SMA patients20. Another issue confounding the feasibility of current SMA therapeutics is their cost effectiveness relative to the high economic burden21,22. While these current therapeutic options have had marked improvements in patient outcomes, the clinical results are still far from an effective cure for SMA. Identifying agents that modulate patient outcomes in addition to current SMA therapies has enormous potential for further maximizing quality of life in these children.
In addition to regulating its promoter, increasing SMN2 expression by modulating the splicing of its pre-mRNA so that a greater proportion of SMN2 mRNAs contain exon 7 has shown great promise in improving motor function and survival in SMA. LDN-76,070 increases FL-SMN mRNA expression and increasing SMN protein levels in SMA patient-derived cells50. High-throughput small molecule screening identified SMN-C1 and its analogue SMN-C3 as potent SMN2 exon 7 splicing enhancers51,52. Branaplam (NVS-SM1) and risdiplam (RG7916) are orally bioavailable analogues of SMN-C1 and RG7800 that promote SMN2 exon 7 inclusion in vivo and improves SMA phenotype53,54. Unfortunately, branaplam and risdiplam both have significant off-target effects. Another analogue of SMN-C3 known as TEC-1 increases exon 7 inclusion and improves SMA phenotype without the off-target effects observed in branaplam and risdiplam55.
Celecoxib activates p38 MAP kinase leading to elevated SMN protein levels and improves SMA phenotype in mice56. Triptolide, a natural product that inhibits the chaperone activity of peroxiredoxin 1, increases SMN protein and improves survival in severe SMA mice57. The calpain inhibitor calpeptin increases SMN protein and improves lifespan of SMA mice58. The terpenoid natural product loganin increases SMN protein in neural tissue as well as increase lifespan in SMNΔ7 SMA mice59. LDN-2014 increases the stability of SMN protein and improves the survival and motor phenotype in two mouse models for SMA60. Translational read-through modulators like geneticin61 and TC007 62,63,64 also increase SMN protein stability and ameliorate the SMA phenotype in mice. ML372 stabilizes SMN protein by blocking its ubiquitination to improve the motor phenotype in SMA mice65. The Ca2+-channel blocker flunarizine increases survival of SMA mice and improves motor function by increasing subnuclear localization of SMN to gems66restoring intranuclear colocalization of SMN and TDP-43 67 and increasing the formation of neuromuscular junctions in SMA mouse muscles68.
While therapies targeting SMN2 have shown great promise in model systems as well as in clinical trials, there is still a strong need for interventions that are independent of SMN2 modulation38. The HDAC inhibitor 4-PBA34 as well as its TSA-linked analogue AR42 37 significantly increase the survival and improve motor function in SMNΔ7 SMA mice without increasing SMN2 expression in vivo. Y-27,632, which inhibits Rho kinase activity, ameliorates the phenotypes of SMA mouse models independent of SMN2 expression69. The JNK inhibitor peptide (D-JNK11) delays motor neuron loss, reduces gliosis and improves muscle innervation in SMA mice70. The effect of D-JNK11 on SMN expression was not measured in this study. Inhibition of autophagy with 3-methyladenine improves survival and motor phenotype in SMA mice within increasing SMN expression71. IPLEX (recombinant human insulin-like growth factor 1) ameliorates motor function and motor neuron loss in SMA mice but has no effect on SMN2 expression72. Prednisolone improves SMA phenotype in mice by upregulating Klf15 expression in muscle73. Inhibition of myostatin activity via activation of activin receptor IIB improves muscle mass in SMA mice74.
Combination therapeutics will be viable strategies for treating SMA especially if these combination treatments increase SMN2 expression at different levels of gene regulation and/or protect vulnerable motor neurons in an SMN2-independent manner. D156844 in combination with either follistatin42 or AR42 (this study) can act additively to improve survival and motor phenotype in SMNΔ7 SMA mice. Increasing FL-SMN transcript levels using two different therapeutics, RG7800 and the splice-correcting oligonucleotide ISS-N1, leads to additive improvements in motor function and survival in SMA mice39. Increasing SMN2 expression by using a combination of a splice-correcting oligonucleotide (MOElv11) and a translational read-through compound (azithromycin monohydrate) additively improves survival and motor phenotype in SMA mice40. The protein arginine methyltransferase inhibitor MS023 works additively with ISS-N1 to improve survival and phenotype in SMA mice75. Coadministration of a SMN2 splice-correcting oligonucleotide and an antisense oligonucleotide targeting the SMA modifier gene neurocalcin D (Ncald) additively improve survival and motor phenotype in SMA mice41. Combination therapies targeting SMN2 expression and muscle degeneration have also demonstrated significant efficacy in SMA. For example, coadministration of the splice-correcting oligonucleotide ISS-N1 and a myostatin propeptide adeno-associated virus vector (AAV8ProMyo) results in significant improvement in SMA mice43. A monoclonal antibody that inhibits myostatin activation (SRK-015) in combination the SMN-C1 increases muscle mass and improves phenotype in SMA mice44.
In summary, we have demonstrated that maternal diet enhances the responsiveness of SMNΔ7 SMA mice to the protective effects of AR-42 (REC-2282), a 4-PBA-tethered TSA analogue. The effect of modifying nutritional status via maternal diet on motor function and viability in SMA mouse models has been shown in other studies76,77. In addition to modulating basal motor phenotype, maternal diet al.so improves the responsiveness of SMA mice to therapeutic interventions like D156844 27, TSA33 and AR-42 (this study). We also show here that the putative small molecule regulator of SMN2 promoter activity D156844 acts synergistically with AR-42. This study exemplifies the potential effectiveness of multi-modal therapeutic strategies for treating SMA, an approach that could also be applied to other early-onset neurodegenerative diseases.
Methods
Animals and ethical statement
SMNΔ7 SMA mice (SMN2+/+; SMNΔ7+/+;mSmn−/−) were generated from male and female carrier mice (SMN2+/+; SMNΔ7+/+;mSmn+/−)45. Since maternal diet influences the survival of SMNΔ7 SMA mice76the breeder mice were provided with ad libitum water and PicoLab20 Mouse diet (#5058; Purina) rodent chow. All animal experiments in this study were performed at Ohio State University and their data were analyzed at Nemours Children’s Hospital Delaware. All experiments were in accordance with the protocols described in the National Institutes of Health Guide for the Care and Use of Animals and were approved by the Ohio State University Institutional Laboratory Animal Care and Use Committee. These studies were completed in accordance with ARRIVE guidelines.
Drugs
D156844 ([5-(1-(2-fluorobenzyl)piperidine-4-ylmethoxy]quinazoline-2,4-diamine dihydrochloride) was obtained from Cure SMA as described previously25. D156844 was dissolved in ddH2O at a concentration of 3 mg/mL. AR42 (Arno Therapeutics, Parsippany, NJ) was dissolved in an aqueous solution containing 0.5% methyl cellulose (molecular mass = 41,000 g/mL; Sigma-Aldrich, St. Louis, MO) and 0.1% Tween-80 (Sigma-Aldrich) at a concentration of 10 mg/mL.
Drug administration
Newborn mice were divided into 4 treatment groups: (1) D156844 (3 mg/kg/d) and AR42 (10 mg/kg/d) (n = 15), (2) AR42 alone (n = 16), (3) D156844 alone (n = 14) and (4) vehicle (0.5% methyl cellulose and 0.1% Tween-80; n = 17). Each group included SMNΔ7 SMA mice as well non-SMA (wild-type or carrier) littermates (n = 17). Mice were dosed daily via oral administration as described previously78. Treatment began at postnatal day 4 (PND04) and continued for the lifetime of each SMA mouse. The body mass of each mouse was determined each day during treatment. The treatment cohorts were not stratified based on sex because there is no significant difference in lifespan between male and female SMNΔ7 SMA mice46 and there are no sex-related differences in the responsiveness of SMNΔ7 SMA mice to D156844 or AR42 26,37.
Phenotype assays
A cohort of SMNΔ7 SMA mice from each treatment group were assayed for changes in righting reflex success and latency, spontaneous locomotor activity and pivoting activity as described previously26,46. All the measures were collected from treated mice at PND07, PND11 and PND14 using Stopwatch+ (Center for Behavioral Neuroscience, Atlanta, GA). The righting reflex was measured by turning each pup onto its back and the time it takes to stably place all four paws on the ground. The cutoff time was 60 s and any pup who was unable to complete this task within the cutoff time received a latency time of 61 s. Directional movement duration was measured as the amount of time each mouse was crawling (PND07) or walking (PND11 and PND14) within the viewing timeframe was collected. For spontaneous locomotor activity, each pup was placed in the center of a gridded (with 28 2.5-cm2 grids) arena and the number of grids crossed in 60 s was counted. For pivoting, each pup was placed in the center of a gridded arena and the number of times the pup turned 90 °C (pivots) during a 60-sec time frame was counted. Between 3 and 5 litters were used for phenotype assessment for each treatment group. To minimize the stress on the pup, the tests for spontaneous locomotor activity and pivoting were conducted simultaneously.
Statistical analysis
Parametric data were expressed as means ± standard errors and were analyzed using one-way ANOVA with a Bonferonni post hoc test. Kaplan-Meier curves were generated from the survival and onset of body mass loss data and tested using the Mantel-Cox log rank test. Kaplan-Meier curves were also generated for other time-to-event phenotype assays (surface righting and spontaneous movement)79. The mice in the treatment groups were also compared against previously published, diet-matched D156844 27 or AR42 37 data. Longitudinal continuous data (body mass curves) for each treatment group were analyzed using generalized estimating equations (GEE)47. Time was included as a covariate. Significance was measured as a Wald χ2 with associated p-value. Statistical significance was defined by a p-value less than or equal to 0.05. All statistical analyses were performed with SPSS v.28.
Data availability
All data pertaining to this study are presented within the manuscript. Further inquiries related to data availability should be directed to M.E.R.B.
References
Crawford, T. O. & Pardo, C. A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 3, 97–110 (1996).
Lefebvre, S. et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165 (1995).
LorsonCL, Hahnen, E., Androphy, E. J. & Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U S A. 96, 6307–6311 (1999).
Monani, U. R. et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183 (1999).
Coovert, D. D. et al. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 6, 1205–1214 (1997).
Lefebvre, S. et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 16, 265–269 (1997).
McAndrew, P. E. et al. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 60, 1411–1422 (1997).
Monani, U. R. et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn-/- mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 9, 333–339 (2000).
Hsieh-Li, H. M. et al. A mouse model for spinal muscular atrophy. Nat. Genet. 24, 66–70 (2000).
Michaud, M. et al. Neuromuscular defects and breathing disorders in a new mouse model of spinal muscular atrophy. Neurobiol. Dis. 38, 125–135 (2010).
Chiriboga, C. A. et al. Results from a phase I study of Nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 86, 890–897 (2016).
Haché, M. et al. Intrathecal injections in children with spinal muscular atrophy: Nusinersen clinical trial experience. J. Child. Neurol. 31, 899–906 (2016).
Finkel, R. S. et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 388, 3017–3026 (2016).
Baranello, G. et al. Risdiplam in type 1 spinal muscular atrophy. N Engl. J. Med. 384, 915–923 (2021).
Mendell, J. R. et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl. J. Med. 377, 1713–1722 (2017).
Gubser Keller, C. et al. An orally available, brain penetrant, small molecule lowers Huntingtin levels by enhancing pseudoexon inclusion. Nat. Commun. 13, 1150 (2022).
Mclean, Z. L. et al. Splice modulators target PMS1 to reduce somatic expansion of the huntington’s disease-associated CAG repeat. Nat. Commun. 15, 3182 (2024).
Coratti, G. et al. Motor function in type 2 and 3 SMA patients treated with nusinersen: A critical review and meta-analysis. Orphanet J. Rare Dis. 16, 430 (2021).
Oskoui, M. et al. Two-year efficacy and safety of Risdiplam in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA). J. Neurol. 270, 2531–2546 (2023).
Fernandes, B. D. et al. Efficacy and safety of onasemnogene abeparvovec for the treatment of patients with spinal muscular atrophy type 1: Systematic review with meta-analysis. PLoS One. 19, e03022860 (2024).
Motta-Santos, A. et al. Cost-effectiveness of technologies for the treatment of spinal muscular atrophy: A systematic review of economic studies. Value Health Reg. Issues. 42, 100985 (2024).
Toro, W. et al. Health care resource utilization and costs for patients with spinal muscular atrophy: Findings from a retrospective US claims database analysis. Adv. Ther. 40, 4589–4605 (2023).
Cherry, J. J. et al. Assays for the identification and prioritization of drug candidates for spinal muscular atrophy. Assay. Drug Dev. Technol. 12, 315–341 (2014).
Jarecki, J. et al. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: Early leads towards a therapeutic for spinal muscular atrophy. Hum. Mol. Genet. 14, 2003–2018 (2005).
Thurmond, J. et al. Synthesis and biological evaluation of novel 2,4-diaminoquinazoline derivatives as SMN2 promoter activators for the potential treatment of spinal muscular atrophy. J. Med. Chem. 51, 449–469 (2008).
Butchbach, M. E. R. et al. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum. Mol. Genet. 19, 454–467 (2010).
Butchbach, M. E. R., Singh, J., Gurney, M. E. & Burghes, A. H. M. The effect of diet on the protective action of D156844 observed in spinal muscular atrophy mice. Exp. Neurol. 256, 1–6 (2014).
Van Meerbeke, J. P. et al. The DcpS inhibitor RG3039 improves motor function in SMA mice. Hum. Mol. Genet. 22, 4074–4083 (2013).
Gogliotti, R. G. et al. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum. Mol. Genet. 22, 4084–4101 (2013).
Avila, A. M. et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Invest. 117, 659–671 (2007).
Andreassi, C. et al. Phenylbutyrate increases SMN expression in vitro: Relevance for treatment of spinal muscular atrophy. Eur. J. Hum. Genet. 12, 59–65 (2004).
Brahe, C. et al. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. Eur. J. Hum. Genet. 13, 256–259 (2005).
Narver, H. L. et al. Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann. Neurol. 64, 465–470 (2008).
Butchbach, M. E. R. et al. Protective effects of butyrate-based compounds on a mouse model for spinal muscular atrophy. Exp. Neurol. 279, 13–26 (2016).
Lu, Q. et al. Zn2+-chelating motif-tethered short-chain fatty acids as a novel class of histone deacetylase inhibitors. J. Med. Chem. 47, 467–474 (2004).
Lu, Q., Wang, D. S., Chen, C. S., Hu, Y. D. & Chen, C. S. Structure-based optimization of phenylbutyrate-derived histone deacetylase inhibitors. J. Med. Chem. 48, 5530–5535 (2005).
Lumpkin, C. J. et al. Evaluation of the orally bioavailable, 4-phenylbutyrate-tethered trichostatin A analogue AR42 in models of spinal muscular atrophy. Sci. Rep. 13, 10374 (2023).
Hensel, N., Kubinski, S. & Claus, P. The need for SMN-independent treatments of spinal muscular atrophy (SMA) to complement SMN-enhancing drugs. Front. Neurol. 11, 45 (2020).
Kray, K. M., McGovern, V. L., Chugh, D., Arnold, W. D. & Burghes, A. H. M. Dual SMN inducing therapies can rescue survival and motor unit function in symptomatic D7 SMA mice. Neurobiol. Dis. 159, 105488 (2021).
Osman, E. Y. et al. Analysis of Azithromycin monohydrate as a single or a combinatorial therapy in a mouse model of severe spinal muscular atrophy. J. Neuromuscul. Dis. 4, 237–249 (2017).
Torres-Benito, L. et al. NCALD antisense oligonucleotide therapy in addition to Nusinersen further ameliorates spinal muscular atrophy in mice. Am. J. Hum. Genet. 105, 221–230 (2019).
Harris, A. W. & Butchbach, M. E. R. The effect of the DcpS inhibitor D156844 on the protective action of follistatin in mice with spinal muscular atrophy. Neuromuscul. Disord. 25, 699–705 (2015).
Zhou, H. et al. Myostatin Inhibition in combination with antisense oligonucleotide therapy improves outcomes in spinal muscular atrophy. J. Cachexia Sarcopenia Muscle. 11, 768–782 (2020).
Long, K. K. et al. Specific Inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum. Mol. Genet. 28, 1076–1089 (2019).
Le, T. T. et al. SMND7, the major product of the centromeric survival motor neuron gene (SMN2), extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 14, 845–857 (2005).
Butchbach, M. E. R., Edwards, J. D. & Burghes, A. H. M. Abnormal motor phenotype in the SMND7 mouse model of spinal muscular atrophy. Neurobiol. Dis. 27, 207–219 (2007).
Liang, K. Y. & Zeber, S. L. Longitudinal data analysis using generalized linear models. Biometrika 73, 13–22 (1986).
Mueller, L., Barrow, P., Jacobsen, B., Ebeling, M. & Weinbauer, G. Reproductive findings in male animals exposed to selective survival of motor neuron 2 (SMN2) gene splicing-modifying agents. Reprod. Toxicol. 118, 108360 (2023).
Theil, D. et al. Neurofilament light chain: a translational safety biomarker for drug-induced peripheral neurotoxicity. Toxicol. Pathol. 51, 135–147 (2023).
Cherry, J. J. et al. Enhancement of SMN protein levels in a mouse model of spinal muscular atrophy using novel drug-like compounds. EMBO Mol. Med. 5, 1103–1118 (2013).
Naryshkin, N. A. et al. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 345, 688–693 (2014).
Zhao, X. et al. Pharmacokinetics, pharmacodynamics and efficacy of a small-molecule SMN2 splicing modifier in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 25, 1885–1899 (2016).
Ratni, H. et al. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J. Med. Chem. 61, 6501–6517 (2018).
Sivaramakrishnan, M. et al. Binding of SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat. Commun. 8, 1476 (2017).
Ando, S. et al. Discovery of a CNS penetrant small molecule SMN2 splicing modulator with improved tolerability for spinal muscular atrophy. Sci. Rep. 10, 17472 (2020).
Farooq, F. et al. Celecoxib increases SMN and survival in a severe spinal muscular atrophy mouse model via p38 pathway activation. Hum. Mol. Genet. 22, 3415–3424 (2013).
Hsu, Y. Y. et al. Triptolide increases transcript and protein levels of survival motor neurons in human SMA fibroblasts and improves survival in SMA-like mice. Br. J. Pharmacol. 166, 1114–1126 (2012).
de la Fuente, S., Sansa, A., Periyakaruppiah, A., Garcera, A. & Soler, R. M. Calpain Inhibition increases SMN protein in spinal cord motoneurons and ameliorates the spinal muscular atrophy phenotype in mice. Mol. Neurobiol. 56, 4414–4427 (2019).
Tseng, Y. T., Chen, C. S., Jong, Y. J., Chang, F. R. & Lo, Y. C. Loganin possess neuroprotective properties, restores SMN protein and activates protein synthesis positive regulator akt/mtor in experimental models of spinal muscular atrophy. Pharmacol. Res. 111, 58–75 (2016).
Osman, E. Y. et al. Intraperitoneal delivery of a novel drug-like compound improves disease severity in severe and intermediate mouse models of spinal muscular atrophy. Sci. Rep. 9, 1633 (2019).
Heier, C. R. & DiDonato, C. J. Translational readthrough by the aminoglycoside geneticin (G418) modulates SMN stability in vitro and improves motor function of SMA mice in vivo. Hum. Mol. Genet. 18, 1310–1322 (2009).
Mattis, V. B., Ebert, A. D., Fosso, M. Y., Chang, C. W. & Lorson, C. L. Delivery of a read-through inducing compound, TC007, lessens the severity of a SMA animal model. Hum. Mol. Genet. 18, 3906–3913 (2009).
Mattis, V. B., Fosso, M. Y., Chang, C. W. & Lorson, C. L. Sucutaneous administration of TC007 reduces disease severity in an animal model of SMA. BMC Neurosci. 10, 142 (2009).
Mattis, V. B., Chang, C. W. T. & Lorson, C. L. Analysis of a read-through promoting compound in a severe mouse model of spinal muscular atrophy. Neurosci. Lett. 525, 72–75 (2012).
Abera, M. B. et al. ML372 blocks SMN ubiquitination and improves spinal muscular atrophy pathology in mice. JCI Insight. 1, e88427 (2016).
Sapaly, D. et al. Small-molecule Flunarizine increases SMN protein in nuclear Cajal bodies and motor function in a mouse model of spinal muscular atrophy. Sci. Rep. 8, 2075 (2018).
Sapaly, D. et al. The small-molecule Flunarizine in spinal muscular atrophy patient fibroblasts impacts on the Gemin components of the SMN complex and TDP43, an RNA-binding protein relevant to motor neuron diseases. Front. Mol. Biosci. 7, 55 (2020).
Delers, P. et al. A link between Agrin signalling and Cav3.2 at the neuromuscular junction in spinal muscular atrophy. Sci. Rep. 12, 18960 (2022).
Bowerman, M., Beauvais, A., Anderson, C. L. & Kothary, R. Rho-kinase inactivation prolongs survival of an intermediate SMA mouse model. Hum. Mol. Genet. 19, 1468–1478 (2010).
Schellino, R., Boido, M., Borsello, T. & Vercelli, A. Pharmacological c-Jun NH2-terminal kinase (JNK) pathway Inhibition reduces severity of spinal muscular atrophy disease in mice. Front. Mol. Neurosci. 11, 308 (2018).
Piras, A. et al. Inhibition of autophagy delays motoneuron degeneration and extends lifespan in a mouse model of spinal muscular atrophy. Cell. Death Dis. 20178, 3223 (2017).
Murdocca, M. et al. IPLEX administration improves motor neuron survival and ameliorates motor functions in a severe mouse model of spinal muscular atrophy. Mol. Med. 18, 1076–1085 (2012).
Walter, L. M. et al. Interventions targeting glucocorticoid-Krüppel-like factor 15-branched-chain amino acid signaling improve disease phenotypes in spinal muscular atrophy mice. EBioMedicine 31, 226–242 (2018).
Liu, M., Hammers, D. W., Barton, E. R. & Sweeney, H. L. Activin receptor type IIB Inhibition improves muscle phenotype and function in a mouse model of spinal muscular atrophy. PLoS One. 11, e0166803 (2016).
Kordala, A. J. et al. PRMT inhibitor promotes SMN2 exon 7 inclusion and synergizes with Nusinersen to rescue SMA mice. EMBO Mol. Med (2023).
Butchbach, M. E. R. et al. Effect of diet on the survival and phenotype of a mouse model for spinal muscular atrophy. Biochem. Biophys. Res. Commun. 391, 835–840 (2010).
Deguise, M. O., Chehade, L., Tierney, A., Beauvais, A. & Kothary, R. Low fat diets increase survival of a mouse model of spinal muscular atrophy. Ann. Clin. Transl Neurol. 6, 2340–2346 (2019).
Butchbach, M. E. R., Edwards, J. D., Schussler, K. R. & Burghes, A. H. M. A novel method for oral delivery of compounds to the neonatal SMND7 model of spinal muscular atrophy. J. Neurosci. Methods. 161, 285–290 (2007).
Jahn-Eimermacher, A., Lasarzik, I. & Raber, J. Statistical analysis of latency outcomes in behavioral experiments. Behav. Brain Res. 221, 271–275 (2011).
Acknowledgements
We would like to thank Dr. Arthur Burghes for providing laboratory space for some of these experiments and Dr. Amanda Hernan for providing guidance on GEE analysis. The study was supported in part by funds from Cure SMA, the Nemours Foundation and the Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (P20GM103464). Cure SMA financially supported and directed the identification and generation of the quinazoline series of compounds, including D156844.
Author information
Authors and Affiliations
Contributions
M.E.R.B. conceived and designed the experiments; A.W.H. and M.E.R.B. performed the experiments; R.C.S. and M.E.R.B. analyzed the data; all authors wrote the paper. All authors have read and approved submission of this work.
Corresponding author
Ethics declarations
Competing interests
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Harris, A.W., Scott, R.C. & Butchbach, M.E.R. The effect of coadministration of D156844 and AR42 (REC-2282) on the survival and motor phenotype of mice with spinal muscular atrophy. Sci Rep 15, 28866 (2025). https://doi.org/10.1038/s41598-025-12194-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-12194-1
