
Abstract
Intervertebral disc degeneration (IDD) severely impacts patients’ quality of life. Elucidating IDD’s molecular mechanisms is crucial for developing effective diagnostics and therapeutics. Integrating multiple IDD gene expression datasets using bioinformatics identified 25 glycosylation-related differentially expressed genes (GRDEGs). We analyzed their biological functions and regulatory networks in IDD using GO, KEGG, GSEA, and WGCNA. An IDD diagnostic LASSO regression model was constructed and validated. Immune cell infiltration analysis using CIBERSORT/ssGSEA divided IDD samples into subtypes based on glycosylation scores, exploring the immune microenvironment’s influence on heterogeneity. We explored regulatory networks involving transcription factors, miRNAs, RNA-binding proteins, and drugs affecting key genes. 25 GRDEGs were identified from 625 glycosylation-related genes (GRGs), and 9 hub genes were further screened by WGCNA to clarify their biological functions and regulatory networks. The diagnostic model based on 7 key genes performed well in the training and validation sets. In addition, Spearman correlation analysis showed that 6 key genes (MAN2B2, MAN1A1, CHI3L1, PLOD2, RAPGEF5, GLA) were significantly associated with immune cell infiltration, including Eosinophils, Dendritic cells, Macrophages M0, and T regulatory cells. Three key genes (MAN2B2, GLA, CHI3L1) significantly affected the differences between high and low glycosylation score subtypes in the immune microenvironment. In addition, regulatory networks, including 49 transcription factors, 32 miRNAs, 116 RNA-binding proteins, and 20 potential drugs, were identified. This study explored the characteristics of glycosylation-related genes in IDD and their potential association with immune infiltration, providing preliminary insights that may inform future research on diagnostic biomarkers and therapeutic targets.
Similar content being viewed by others


Introduction
Intervertebral disc degeneration (IDD) is a common chronic degenerative disease and one of the main causes of low back pain1. Epidemiological studies have shown that the global prevalence of IDD ranges from 50% to 90%, significantly affecting patients’ quality of life2. Although current evidence-based medicine has identified IDD as a result of multiple factors, including genetics, trauma, inflammation, lifestyle, and aging, the pathogenic processes involved in IDD development remain unclear3. In recent years, the development of genomics and bioinformatics technologies has provided new ideas for in-depth exploration of the molecular mechanisms of IDD4.
Glycosylation, an important modification that occurs after proteins and lipids have been translated, is vital for many biological functions, including cell adhesion and the transmission of signals5,6. Previous studies have indicated that the occurrence of multiple degenerative diseases, such as osteoarthritis and Alzheimer’s disease, is closely related to abnormal glycosylation7,8. However, the impact of glycosylation-related genes (GRGs) on the progression of IDD is still not well understood. Therefore, this study aims to screen and validate IDD-related glycosylation genes through bioinformatic methods, providing new ideas for understanding the molecular mechanisms of IDD and exploring potential therapeutic targets.
Currently, multiple studies9,10,11,12 have reported biological markers and pathogenic mechanisms related to IDD, but most are limited to the protein level, and the regulatory mechanisms at the gene level have not been thoroughly explored. In recent years, with the development of omics technologies, researchers have begun to focus on the screening and functional analysis of IDD-related genes13,14. However, these studies mainly concentrate on the identification of differentially expressed genes (DEGs), with relatively less research on their functions and regulatory mechanisms. Moreover, IDD is a complex disease involving multiple pathological processes, and single omics analysis may not fully reveal its pathogenesis.
Zhu et al. demonstrated the significant contribution of genes related to mitochondrial dysfunction to the advancement of IDD through an extensive bioinformatics investigation15. Another study utilized genome-wide analysis of DNA methylation profiles to identify differentially methylated sites associated with human IDD16. However, there are few reports on the use of multi-omics methods to study IDD-related glycosylation genes.
This study intends to integrate multiple IDD gene expression profile datasets, taking GRGs as a starting point, and comprehensively apply various bioinformatic methods such as differential analysis, Weighted Gene Co-Expression Network Analysis (WGCNA), and Gene Set Enrichment Analysis (GSEA) to analyze the molecular mechanisms of IDD development from the perspectives of gene co-expression networks and pathway enrichment, screening potential markers and therapeutic targets. At the same time, a diagnostic model for IDD based on Least Absolute Shrinkage and Selection Operator (LASSO) regression was developed and tested for reliability and accuracy. The study also investigated the correlation between critical genes and immune infiltration using CIBERSORT and single sample gene set enrichment analysis (ssGSEA). By categorizing IDD samples into immune subtypes, the research highlighted the important role of immune microenvironment changes in IDD heterogeneity. Additionally, the study delved into the regulatory networks of transcription factors, miRNAs, RNA-binding proteins, and drugs on essential genes.This research is crucial for elucidating IDD pathophysiology and guiding the enhancement of clinical diagnosis and treatment strategies. Furthermore, the research may serve as a valuable reference for exploring mechanisms of other degenerative diseases.
Materials and methods
Data collection and downloading
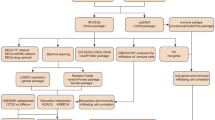
Figure 1 showed the workflow chart of the present study. The IDD datasets GSE3409517, GSE7036218, and GSE14738319 were downloaded from the GEO database20 using the R package GEO query21. The samples in these datasets were all human, derived from intervertebral disc nucleus pulposus tissue. The datasets GSE34095, GSE70362, and GSE147383 used the chip platforms GPL96, GPL17810, and GPL570, respectively. GSE34095 included 3 IDD samples and 3 control samples; GSE70362 included 16 IDD samples and 8 control samples; GSE147383 included 2 IDD samples and 2 control samples. GRGs were collected using the GeneCards22 and MSigDB23 databases and published literature24, obtaining a total of 625 unique genes (Supplementary Table S1). The R package sva24 was used for batch effect removal, and the integrated dataset contained 21 IDD samples and 13 control samples (see Table 1 for details). The R package limma24 was used for data normalization and batch effect removal, and principal component analysis (PCA)25 was performed to verify the batch effect removal (Fig. 2).

Technology roadmap. IDD, Intervertebral Disc Degeneration; DEG, Differentially Expressed Genes; GRGs, Glycosylation-Related Genes; GRDEGs, Glycosylation Related Differentially Expressed Genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; GSEA, Gene Set Enrichment Analysis; ssGSEA, Single-Sample Gene-Set Enrichment Analysis; SVM, Support Vector Machine; LASSO, Least Absolute Shrinkage and Selection Operator; PPI Network, Protein-Protein Interaction Network; TF, Transcription Factor; RBP, RNA-Binding Protein.

Batch effects removal of combined datasets. (A) Boxplots of GEO Datasets (Combined Datasets) distribution before batch removal. (B) Post-batch integrated GEO Datasets (Combined Datasets) distribution boxplots. (C) PCA plot of the datasets before debatching. (D) Go to the PCA map of the Combined GEO Datasets after batch processing. IDD, Intervertebral Disc Degeneration; PCA, Principal Component Analysis. The intervertebral disc degeneration (IDD) dataset GSE34095 is blue, the IDD dataset GSE70362 is red, and the IDD dataset GSE147383 is yellow.
IDD-related glycosylation-related differentially expressed genes
In the integrated GEO dataset, samples were categorized into the IDD group and the Control group. Differential gene analysis was performed using the R package limma, with the threshold set at |logFC| > 0.3 and p-value 0.3 were considered upregulated, while those with logFC < – 0.3 were deemed downregulated. The results of the analysis were visualized using a volcano plot generated with the R package ggplot2. Intersection of all DEGs with GRGs yielded IDD-related glycosylation-related differentially expressed genes (GRDEGs). The expression levels of GRDEGs were compared between groups using the Mann-Whitney U test, and a heatmap was constructed using the R package pheatmap. Chromosomal locations of GRDEGs were visualized using the R package RCircos26.
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis
GO analysis, as referenced by27, is a technique employed for conducting functional enrichment studies across biological processes (BP), cellular components (CC), and molecular functions (MF). KEGG was also utilized as a database delineating information on genomes, biological pathways, diseases, and drugs28,29,30. The R package clusterProfiler facilitated the GO and KEGG enrichment analysis of GRDEGs28. To ensure statistical significance, the Benjamini-Hochberg method was employed for p-value correction, with screening criteria set at adj. p < 0.05 and FDR < 0.25.
GSEA between IDD and control groups
GSEA31 was conducted to assess the distribution of predefined gene sets within the gene table and their impact on the phenotype. The genes from the integrated GEO dataset were arranged based on logFC values, and GSEA analysis was performed using the R package clusterProfiler. The analysis was carried out with specific parameters, including a seed of 2022, 1000 permutations, and gene set sizes ranging from 10 to 500 genes. The gene set c2.all.v2022.1.Hs.symbols.GMT [Curated/Pathway] (6449) from the MSigDB32 database was utilized for the analysis. The Benjamini-Hochberg correction method was applied with an adjusted p-value < 0.05 and false discovery rate (FDR) < 0.25 for significance.
Weighted gene co-expression network analysis
Using the R package WGCNA33,34, weighted correlation coefficients between genes were initially computed to build a scale-free network. Initially, pairwise correlation coefficients were computed between all genes, with weighted correlation values applied to establish a scale-free network topology for gene connectivity. A hierarchical clustering tree was then constructed based on inter-gene correlations, where distinct branches represented gene modules (color-coded), followed by module significance assessment. For the integrated GEO datasets, the variance was calculated across all genes to select the top 3000 high-variance genes, with parameters set to minimum module size = 80 and optimal soft-thresholding power = 5. Module-trait correlations between IDD and Control groups were measured, defining all genes within each module as module eigengenes. Modules exhibiting |r| > 0.40 were screened, and their constituent genes intersected with Glycosylation-Related Differentially Expressed Genes (GRDEGs) to generate Venn diagrams, with all intersecting genes from qualified modules designated hub genes. Finally, Spearman correlation analysis was performed on hub gene expression profiles within the integrated GEO dataset. Resulting correlation matrices were visualized using R packages igraph and ggraph with defined strength thresholds: |r| < 0.3 (Weak/Non-significant), 0.3 ≤ |r| < 0.5 (Weak), 0.5 ≤ |r| < 0.8 (Moderate), and |r| ≥ 0.8 (Strong).
Construction of IDD diagnostic model
To construct the IDD diagnostic model, logistic regression analysis was conducted on hub genes to analyze the association between IDD and Control. Genes with a p-value < 0.05 were identified as GRDEGs. These genes were then used to construct a forest plot displaying the expression levels in the logistic regression model. Subsequently, a model was created utilizing the Support Vector Machine (SVM) algorithm, focusing on genes with the highest accuracy35. Finally, LASSO regression analysis was performed using the R package glmnet36, setting the seed to 500 and the family to “binomial” to reduce overfitting and improve generalization ability. The LASSO results were depicted in the diagnostic model and variable trajectory plot. The hub genes in the results were defined as key genes. The risk score formula based on the LASSO regression coefficients was calculated as follows:
Validation of IDD diagnostic model
Based on the LASSO regression results, a nomogram37 was drawn using the R package rms to display the relationships among hub genes. Calibration plots were used to evaluate the accuracy and discrimination of IDD diagnostic models. Decision curve analysis (DCA)38 was performed using the ggDCA package in R to assess the clinical utility of the predictive model. In addition, ROC curves based on the LASSO risk score and key genes were plotted using the R package pROC, and AUC values were calculated to assess the diagnostic performance. An AUC close to 1 indicated high diagnostic accuracy. Based on the risk score, samples with IDD were categorized into high-risk and low-risk groups. The Mann-Whitney U test was employed to assess the expression variances of key genes between these groups, with the outcomes presented in group comparison plots.
Immune infiltration analysis between IDD and control groups
The CIBERSORT algorithm39 and LM22 signature gene matrix were employed to analyze the transcriptome expression matrix of the combined GEO dataset samples. This process estimated the composition and abundance of immune cells, with a focus on data displaying immune cell enrichment scores above zero. Stacked bar plots illustrating the proportions of LM22 immune cells in both the IDD and Control groups were generated using the R package ggplot2. Additionally, correlations between immune cells and key genes, as well as correlations among immune cells themselves, were calculated using the Spearman algorithm. Correlation heatmaps were then created using the R packages pheatmap and ggplot2. Based on the correlation heatmap, the four key genes with the strongest correlations with immune cells were selected and further displayed through scatter plots.
Construction of high and low glycosylation score groups
The ssGSEA algorithm40 was utilized with the R package GSVA to compute glycosylation scores (Gs) for all samples in the integrated GEO dataset. These scores were then used to categorize IDD samples into high (HighScore) and low (LowScore) score groups. ROC curves of glycosylation scores and key genes were generated using the R package pROC to assess their diagnostic performance for IDD. The AUC values varied from 0.5 to 1, where 0.5 ~ 0.7 indicated low accuracy, 0.7 ~ 0.9 indicated moderate accuracy, and above 0.9 indicated high accuracy. Subsequently, the Mann-Whitney U test was employed to compare the expression variances of key genes between the high and low score groups, and the outcomes were illustrated through group comparison plots.
Immune infiltration analysis between high and low glycosylation score groups
Utilizing the CIBERSORT algorithm39 in conjunction with the LM22 signature gene matrix, IDD samples within the combined GEO dataset were analyzed to estimate the composition and abundance of immune cells. The analysis included only data with immune cell enrichment scores above zero. The results were visualized using stacked bar plots drawn with the R package ggplot2, showing the proportions of LM22 immune cells in the HighScore and LowScore groups. The Spearman algorithm was conducted to examine the relationship between hub genes and the abundance of immune cell infiltration, retaining results with p-value < 0.05, and a correlation heatmap was drawn using ggplot2. Finally, immune cells significantly correlated with key genes were selected, and lollipop plots were drawn using ggplot2 to further display these relationships.
Immune infiltration analysis and consensus clustering
ssGSEA40 quantitatively evaluated the infiltration abundance of immune cells in IDD samples from the comprehensive GEO dataset, encompassing diverse immune cell subtypes including activated CD8 T cells and dendritic cells. The IDD samples were subjected to analysis using consensus clustering41 based on the immune cell infiltration matrix, with the R package ConsensusClusterPlus42. The number of clusters ranged from 2 to 9, and the clustering process was repeated 50 times with 80% of the sample size. Furthermore, the analysis included examination of the expression variances of key genes across different IDD subtypes, as well as the expression disparities of immune cells within the various IDD subtypes. Finally, the Spearman algorithm was employed to explore the correlation between key genes and immune cells, with the resulting correlations visualized in heatmaps.
Construction of protein-protein interaction network
Based on central genes, the STRING 12.0 database was used to construct a protein-protein interaction (PPI) network of key genes43, selecting genes with a minimum interaction coefficient greater than 0.150 for in-depth analysis. Meanwhile, the GeneMANIA 3.5.1 database44 was utilized to predict and analyze functionally similar genes of key genes, further constructing a PPI network to assist in gene function analysis and prediction.
Construction of regulatory networks
Transcription factors (TFs) regulate gene expression by interacting with specific key genes. The regulatory effects of TFs on key genes were analyzed using the ChIPBase45 and hTFtarget46 databases, while the mRNA-TF regulatory network was visualized using Cytoscape 3.10.147. Additionally, the relationship between key genes and miRNAs was examined through the StarBase v3.0 database48, and the mRNA-miRNA regulatory network was visualized. Predictions for target RNA-binding proteins (RBPs) of key genes were made using the same database, and the mRNA-RBP regulatory network was visualized49. Finally, drug targets of key genes were predicted using the CTD database50, and the mRNA-Drug regulatory network was visualized through Cytoscape, completing the network construction.
Statistical analysis
All data processing and analysis in this study were conducted using R software (Version 4.3.1). In cases where continuous variables were compared between two groups, the independent Student’s T-test was utilized for normally distributed variables, while the Mann-Whitney U test (Wilcoxon Rank Sum Test) was employed for non-normally distributed variables. For comparisons involving three or more groups, the Kruskal-Wallis test was applied. Spearman correlation analysis was used to calculate correlation coefficients between different molecules. Unless otherwise specified, all statistical p-values were two-sided, with a significance level set at p < 0.05.
Results
Intervertebral disc degeneration-related glycosylation-related differentially expressed genes
Utilizing the R package limma for differential gene analysis, a total of 559 DEGs were screened, including 282 upregulated genes and 277 downregulated genes. A volcano plot was drawn based on the differential analysis results of this dataset (Fig. 3A).
According to the differential analysis method, DEGs and GRGs were obtained, and a Venn diagram was drawn by taking their intersection (Fig. 3B), yielding a total of 25 GRDEGs: IGFBP3, MUC1, MAN2B2, ST6GALNAC2, ST8SIA1, HEXA, CNIH3, PIGT, PTGDS, MAN1A1, DPAGT1, GALNT7, SERPINA1, PDPN, EDEM3, RAPGEF5, C1GALT1C1, TSPAN1, GLA, TLR4, PLOD2, ATP6AP2, GALNT3, CHI3L1, and THBS1. Detailed information on GRDEGs is listed in Table 2. Based on the intersection results, the locations of the 25 GRDEGs on human chromosomes were analyzed using the R package RCircos, and a chromosomal location map was drawn (Fig. 3C). The chromosomal location map shows that the 25 GRDEGs are located on chromosomes 1, 2, 3, 4, 6, 7, 9, 11, 12, 14, 15, 17, 20, and X.
A simple value heatmap (Fig. 3D) and group comparison plot (Fig. 3E) were drawn using the R package ggplot2 to display the analysis results. All 25 GRDEGs showed significant differences between the different sample groups. The genes IGFBP3, ST6GALNAC2, CNIH3, PTGDS, MAN1A1, DPAGT1, GALNT7, PDPN, EDEM3, C1GALT1C1, GLA, TLR4, PLOD2, ATP6AP2, CHI3L1, and THBS1 were significantly up regulated in the IDD group, while the genes MUC1, MAN2B2, ST8SIA1, HEXA, PIGT, SERPINA1, RAPGEF5, TSPAN1, and GALNT3 were abundantly expressed in the Control group.

Differential gene expression analysis. (A) Volcano plot of DEGs analysis between IDD and Control in the integrated GEO dataset, with GRDEGs marked. (B) Venn diagram of DEGs and GRGs in the integrated GEO dataset. (C) Chromosomal location map of GRDEGs. (D, E) Simple value heatmap (D) and group comparison plot (E) of GRDEGs expression levels between IDD and Control groups in the integrated GEO dataset. IDD, Intervertebral Disc Degeneration; DEGs, Differentially Expressed Genes; GRGs, Glycosylation-Related Genes; GRDEGs, Glycosylation-Related Differentially Expressed Genes. *Represents p-value < 0.05, indicating statistical significance; ** represents p-value < 0.01, indicating high statistical significance; *** represents p-value < 0.01, indicating extreme statistical significance. In the grouping, red represents IDD and blue represents Control; in the simple value heatmap, red represents high expression and blue represents low expression.
GO and KEGG enrichment analysis
The detailed results are shown in Supplementary Table S2. The results indicate that 12 GRDEGs are mainly enriched in protein glycosylation (BP), lysosomal lumen (CC), and hydrolase activity (MF) in IDD (Fig. 4A–C). They are also enriched in the mucin type O-glycan biosynthesis pathway (KEGG) (Fig. 4D).

GO and KEGG enrichment analysis for GRDEGs. (A–D) Bar plot display of Gene Ontology (GO) biological process (BP), cellular component (CC), molecular function (MF), and pathway (KEGG) enrichment analysis results of GRDEGs: BP (A), CC (B), MF (C), and KEGG (D). The horizontal axis represents GO terms and KEGG terms. GRDEGs, Glycosylation-Related Differentially Expressed Genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; BP, Biological Process; CC, Cell Component; MF, Molecular Function. The screening criteria for Gene Ontology (GO) and pathway (KEGG) enrichment analysis were adj.p < 0.05 and FDR value (q value) < 0.25, and the p-value correction method was Benjamini-Hochberg (BH).
GSEA between IDD and control groups
The GSEA results (Fig. 5A) are shown in Table 3. The results demonstrate that all genes in the integrated GEO dataset are markedly elevated in iron metabolism in placenta (Fig. 5B), adaptation to hypoxia down (Fig. 5C), apoptosis by serum deprivation up (Fig. 5D), integrated TGF-β EMT up (Fig. 5E), and other biologically relevant functions and signaling pathways.

GSEA for intervertebral disc degeneration between IDD and control groups. (A) The gene set enrichment analysis (GSEA) 7 biological functions enrichment plot display of the Combined GEO Datasets. (B–E) Gene set enrichment analysis (GSEA) showed that all genes were significantly enriched in Hypoxia Dn (B), Apoptosis By CDKN1A Via TP53 (C), Emt Breast Tumor Dn (D), and apoptosis by CDKN1A via TP53 (C). Circadian Rhythm Genes (E). IDD, Intervertebral Disc Degeneration; GSEA, Gene Set Enrichment Analysis. The screening criteria of gene set enrichment analysis (GSEA) were adj.p < 0.05 and FDR value (q value) < 0.25, and the p value correction method was Benjamini-Hochberg (BH).
Weighted gene co-expression network analysis
The WGCNA results (Fig. 6A) show that the top 3,000 genes with the highest variance were clustered and annotated with grouping information through a clustering tree (Fig. 6C). The genes were aggregated into 12 modules (Fig. 6B). Using |r value| > 0.40 as the criterion for modules, two modules were selected for subsequent analysis: MEpink and MEgreen. The 25 GRDEGs were intersected with the genes contained in the two modules, and a Venn diagram was drawn (Fig. 6D), yielding a total of 9 hub genes: IGFBP3, MAN2B2, PTGDS, MAN1A1, SERPINA1, RAPGEF5, GLA, PLOD2, and CHI3L1.
Finally, the correlation heatmap of hub gene expression levels (Fig. 6E) shows that the gene SERPINA1 has the strongest significant positive correlation with the gene MAN2B2 (r value = 0.617, p-value < 0.001), while the gene GLA exhibits the most robust and significant negative correlation with the gene MAN2B2 (r value = – 0.679, p-value < 0.001).

WGCNA for combined datasets. (A) Display of the scale-free network with the optimal soft threshold in weighted gene co-expression network analysis (WGCNA). The left plot shows the optimal soft threshold, and the right plot shows the network connectivity under different soft thresholds. (B) Display of the correlation analysis results between clustering modules of the top 3,000 genes with the highest variance and the Control and IDD groups. (C) Display of the module aggregation results of the top 3,000 genes with the highest variance. (D) Display of the Venn diagram of 25 GRDEGs and genes contained in the MEpink and MEgreen modules. (E) Correlation heatmap of expression levels between hub genes. IDD, Intervertebral Disc Degeneration; WGCNA, Weighted Gene Co-Expression Network Analysis; GRDEGs, Glycosylation-Related Differentially Expressed Genes. *Represents p-value < 0.05, indicating statistical significance; ** represents p-value < 0.01, indicating high statistical significance; *** represents p-value < 0.01, indicating extreme statistical significance. The absolute value of the correlation coefficient (r value) below 0.3 is weak or uncorrelated, between 0.3 and 0.5 is weakly correlated, between 0.5 and 0.8 is moderately correlated, and above 0.8 is strongly correlated. Red represents positive correlation and blue represents negative correlation.
Construction of IDD diagnostic model
The logistic regression model was constructed using the 9 hub genes and displayed through a forest plot (Fig. 7A). Detailed information is shown in Table 4. The results indicate that all 9 hub genes have statistical significance in the logistic regression model (p-value < 0.05). Next, an SVM model was constructed based on the 9 hub genes and the SVM algorithm, obtaining the genes with the lowest error rate (Fig. 7B) and the highest accuracy (Fig. 7C). The results show that when the number of genes is 8, the accuracy of the SVM model is the highest. These 8 hub genes are MAN2B2, IGFBP3, MAN1A1, CHI3L1, PLOD2, RAPGEF5, GLA, and PTGDS.

Diagnostic model of intervertebral disc degeneration. (A) Forest Plot of the nine hub genes included in the Logistic regression model in the diagnostic model of intervertebral disc degeneration (IDD). (B, C) Visualization of the number of genes with the lowest error rate (B) and the number of genes with the highest accuracy (C) obtained by the SVM algorithm. (D, E) Diagnostic model plot (D) and variable trajectory plot (E) of LASSO regression model. IDD, Intervertebral Disc Degeneration; SVM, Support Vector Machine; LASSO, Least Absolute Shrinkage and Selection Operator.
A LASSO regression analysis was conducted using the 8 hub genes from the SVM model to develop an IDD diagnostic model. Visual representations of the LASSO regression model diagram (Fig. 7D) and the LASSO variable trajectory diagram ((Fig. 7E) were created. The analysis identified 7 key genes in the LASSO regression model: MAN2B2, IGFBP3, MAN1A1, CHI3L1, PLOD2, RAPGEF5, and GLA.
Validation of IDD diagnostic model
To further substantiate the value of the IDD diagnostic model, a nomogram was drawn based on the key genes to display the relationships among key genes in the integrated GEO dataset (Fig. 8A). The findings suggest that the expression level of the critical gene MAN2B2 is significantly more useful for the IDD diagnostic model compared to other variables. In contrast, the expression level of GLA has notably less utility for the IDD diagnostic model compared to other variables.
The calibration curve plot of the IDD diagnostic model indicates that the calibration line, represented by the dashed line, slightly deviates from the diagonal line of the ideal model but closely coincides with it (Fig. 8B). The DCA plot demonstrates that the model line consistently outperforms All positive and All negative within a specific range, indicating that the model provides greater benefits and superior performance (Fig. 8C). Furthermore, the ROC curve reveals that the risk score’s expression level in the integrated GEO dataset exhibits high accuracy (AUC > 0.9) across different groups (Fig. 8D). Simultaneously, drawing an ROC curve based on the expression levels of 7 critical genes in the integrated GEO dataset shows moderate accuracy (0.7 < AUC < 0.9) between different groups in this dataset (Fig. 8E). The formula for calculating risk score (Eq. 1) is as follows:
Subsequently, group comparison plots were utilized to explore the expression differences of critical genes in IDD. The results of the differential analysis of the expression levels of the 7 essential genes in the HighRisk and LowRisk groups of IDD are displayed in Fig. 8F. It was found that the essential genes MAN2B2, IGFBP3, and GLA exhibit statistically significant differences in expression levels between the HighRisk and LowRisk groups of IDD (p-value < 0.05). Specifically, IGFBP3 and GLA are highly expressed in the high-risk group, while MAN2B2 is highly expressed in the low-risk group.

Diagnostic and validation analysis of intervertebral disc degeneration. (A) Nomogram of key genes in the IDD diagnostic model in the integrated GEO dataset. (B) Calibration curve plot of the IDD diagnostic model based on key genes in the integrated GEO dataset. (C) DCA plot of the IDD diagnostic model based on the risk score (RiskScore) in the integrated GEO dataset. (D) ROC curve of the risk score (RiskScore) in the integrated GEO dataset. (E) ROC curve of key genes in the integrated GEO dataset. (F) Group comparison plot of key genes in the HighRisk and LowRisk groups of IDD. The vertical axis of the calibration curve plot represents the net benefit, and the horizontal axis represents the probability threshold or threshold probability. IDD, Intervertebral Disc Degeneration; DCA, Decision Curve Analysis; ROC, Receiver Operating Characteristic; AUC, Area Under the Curve; CI, Confidence Interval; TPR, True Positive Rate; FPR, False Positive Rate. ns represents p-value ≥ 0.05, indicating no statistical significance; * represents p-value < 0.05, indicating statistical significance; ** represents p-value < 0.01, indicating high statistical significance. When AUC > 0.5, it indicates a trend of the molecule’s expression promoting the occurrence of the event, and the closer the AUC is to 1, the better the diagnostic effect. AUC between 0.5 and 0.7 indicates low accuracy, AUC between 0.7 and 0.9 indicates moderate accuracy, and AUC above 0.9 indicates high accuracy. Red represents the HighRisk group and blue represents the LowRisk group.
Immune infiltration analysis between IDD and control groups
The CIBERSORT algorithm was used to calculate the immune infiltration abundance in the IDD and Control groups. The results show that 17 immune cell types are enriched in IDD samples (Fig. 9A). According to the correlation heatmap of immune cell infiltration abundance (Fig. 9B), follicular helper T cells have the strongest significant positive correlation with activated dendritic cells (r value = 0.556, p-value < 0.001), while activated NK cells have the strongest significant negative correlation with activated mast cells (r value = – 0.584, p-value < 0.001). According to the correlation heatmap between key genes and immune cell infiltration abundance (Fig. 9C), in IDD samples, the gene GLA has a significant positive correlation with eosinophils (r value = 0.581, p-value < 0.001), the gene PLOD2 has a significant positive correlation with M0 macrophages (r value = 0.544, p-value < 0.001), the gene GLA has a significant negative correlation with activated dendritic cells (r value = – 0.438, p-value < 0.01), and the gene RAPGEF5 has a significant negative correlation with regulatory T cells (Tregs) (r value = – 0.429, p-value < 0.05). Finally, scatter plots (Fig. 9D–G) were drawn to further display the correlations between the gene GLA and eosinophils (Fig. 9D), the gene PLOD2 and M0 macrophages (Fig. 9E), the gene GLA and activated dendritic cells (Fig. 9F), and the gene RAPGEF5 and regulatory T cells (Tregs) (Fig. 9G).

Immunoinfiltration analysis between IDD and control groups (CIBERSORT). (A) Stacked bar plot of the proportions of LM22 immune cells in the integrated GEO dataset. (B) Correlation heatmap of immune cell infiltration abundance in the integrated GEO dataset. (C) Correlation heatmap between immune cell infiltration abundance and key genes in the integrated GEO dataset. (D) Scatter plot of the correlation between the gene GLA and eosinophils. (E) Scatter plot of the correlation between the gene PLOD2 and M0 macrophages. (F) Scatter plot of the correlation between the gene GLA and activated dendritic cells. (G) Scatter plot of the correlation between the gene RAPGEF5 and regulatory T cells (Tregs). IDD, Intervertebral Disc Degeneration. *Represents p-value < 0.01, indicating statistical significance; ** represents p-value < 0.01, indicating high statistical significance; *** represents p-value < 0.001, indicating extreme statistical significance. The absolute value of the correlation coefficient (r value) below 0.3 is weak or uncorrelated, between 0.3 and 0.5 is weakly correlated, between 0.5 and 0.8 is moderately correlated, and above 0.8 is strongly correlated. In the grouping, red represents the IDD group and blue represents the Control group. In the correlation heatmap, red represents positive correlation and blue represents negative correlation, with the color depth representing the strength of the correlation.
Immune infiltration analysis between high and low glycosylation score groups
Based on the expression levels of the 7 critical genes in the integrated GEO dataset, glycosylation scores (Gs) for all samples were calculated using the ssGSEA algorithm. Additionally, ROC curves were generated using the R package pROC based on the Gs in the integrated GEO dataset. The ROC curves (Fig. 10A) indicate that Gs expression level demonstrates moderate accuracy (0.7 < AUC < 0.9) between different groups in the integrated GEO dataset. Furthermore, a group comparison plot was created using the R package ggplot2 (Fig. 10B), revealing highly significant statistical differences in Gs between IDD and Control groups within the integrated GEO dataset (p-value < 0.001). Subsequently, based on median Gs values of IDD samples, they were divided into high (HighScore) and low (LowScore) glycosylation score groups. Differential analysis of genes in these high and low-score groups was conducted using the R package limma, with results displayed through group comparison plots (Fig. 10C). These results demonstrate statistically significant differences in expression levels of key genes CHI3L1, MAN1A1, and PLOD2 between HighScore and LowScore groups of IDD (p-value < 0.05), with all three being highly expressed in the low score group.
Subsequently, the results of immune infiltration analysis were used to create a stacked bar plot illustrating the proportions of immune cells in the integrated GEO dataset (Fig. 10D). Additionally, a correlation heatmap was generated to visually represent the correlation between critical genes and immune cell infiltration abundance (Fig. 10E). The findings indicate that in IDD samples, the gene PLOD2 exhibits a strong significant positive correlation with M0 macrophages (r-value = 0.736, p-value < 0.001). Conversely, the gene MAN2B2 demonstrates a strong significant negative correlation with eosinophils (r-value = -0.534, p-value < 0.05). The key genes with significant correlations with eosinophils are the most numerous. The key genes MAN2B2, GLA, and CHI3L1 have significant correlations with eosinophils, and lollipop plots (Fig. 10F) were drawn to further display the correlations between the genes MAN2B2, GLA, CHI3L1, and eosinophils.

Immunoinfiltration analysis between high and low score groups (CIBERSORT). (A) ROC curve of the glycosylation score (Gs) in the integrated GEO dataset. (B) Group comparison plot of the glycosylation score (Gs) between IDD and Control groups in the integrated GEO dataset (Combined Datasets). (C) Group comparison plot of key genes in the HighScore and LowScore groups of IDD samples. (D) Stacked bar plot of the proportions of LM22 immune cells in IDD samples. (E) Correlation heatmap between immune cell infiltration abundance and key genes in IDD samples. (F) Lollipop plot of the correlations between key genes MAN2B2, GLA, CHI3L1, and eosinophils. A positive correlation coefficient indicates a positive correlation between the two variables, while a negative correlation coefficient indicates a negative correlation between the two variables. IDD, Intervertebral Disc Degeneration. ns represents p-value ≥ 0.05, indicating no statistical significance; * represents p-value < 0.01, indicating statistical significance; ** represents p-value < 0.01, indicating high statistical significance; *** represents p-value < 0.001, indicating extreme statistical significance. The absolute value of the correlation coefficient (r value) below 0.3 is weak or uncorrelated, between 0.3 and 0.5 is weakly correlated, between 0.5 and 0.8 is moderately correlated, and above 0.8 is strongly correlated. In the group comparison plot (B), red represents the IDD group and blue represents the Control group; in the group comparison plot (C) and stacked bar plot (D), red represents the HighScore group and blue represents the LowScore group; in the correlation heatmap, red represents positive correlation and blue represents negative correlation, with the color depth representing the strength of the correlation.
Immune infiltration analysis and consensus clustering based on immune characteristics of IDD samples
Using the k-means unsupervised clustering method, based on the infiltration levels of 28 immune cell types, all samples were clustered into two IDD subtypes (Fig. 11A): subtype 1 (Cluster1) and subtype 2 (Cluster2). The PCA results (Fig. 11B) show that in the reduced dimensionality space, there is a clear and distinct boundary between the two groups of samples, indicating good clustering performance.
Subsequently, the expression differences of key genes between subtype 1 (Cluster1) and subtype 2 (Cluster2) are shown in a volcano plot (Fig. 11C). The results indicate that the genes MAN1A1 and IGFBP3 are highly expressed in subtype 2. At the same time, the group comparison of 28 immune cell types between subtype 1 and subtype 2 (Fig. 11D) demonstrates that 12 immune cell types are enriched in IDD samples and have significant statistical differences between subtype 1 and subtype 2: activated CD4 T cells, activated dendritic cells, CD56dim natural killer cells, central memory CD4 T cells, effector memory CD8 T cells, eosinophils, MDSCs, memory B cells, plasmacytoid dendritic cells, regulatory T cells, T follicular helper cells, and type 1 T helper cells. Subsequently, a simple value heatmap (Fig. 11E) was used to further display the differences in the infiltration levels of the 12 immune cell types between subtype 1 and subtype 2 in IDD samples.
Finally, based on the correlation heatmap between essential genes and immune cell infiltration abundance (Fig. 11F), it was found that in IDD samples, the gene MAN2B2 exhibits a strong significant positive correlation with memory B cells (r-value = 0.649, p-value < 0.01). Conversely, the gene GLA shows a strong significant negative correlation with memory B cells (r-value = – 0.645, p-value < 0.01).

Immunoinfiltration analysis between Cluster1 and Cluster2 groups (ssGSEA). (A) Consensus clustering results of IDD samples from the integrated GEO dataset based on the infiltration levels of 28 immune cell types calculated by the ssGSEA algorithm. (B) PCA plot of the two IDD disease subtypes. (C) Volcano plot of the differential analysis results in IDD subtypes, with key genes marked. (D) Group comparison plot of the infiltration levels of 28 immune cell types between the two IDD subtype groups. (E) Simple value heatmap of the infiltration levels of the selected 12 immune cell types between the two IDD subtype groups. (F) Correlation heatmap between key genes and the infiltration abundance of 12 immune cell types. IDD, Intervertebral Disc Degeneration; PCA, Principal Component Analysis. ns represents p-value ≥ 0.05, indicating no statistical significance; * represents p-value < 0.01, indicating statistical significance; ** represents p-value < 0.01, indicating high statistical significance. In the grouping, blue represents subtype 1 (Cluster1) and red represents subtype 2 (Cluster2); in the simple value heatmap, red represents high expression and blue represents low expression; in the correlation heatmap, red represents positive correlation and blue represents negative correlation, with the color depth representing the strength of the correlation.
Construction of PPI network
First, the PPI network results (Fig. 12A) indicate that 6 key genes are related: MAN2B2, IGFBP3, MAN1A1, CHI3L1, PLOD2, and GLA. Then, use the Cytoscape software to draw the PPI network of these 6 key genes (Fig. 12B). Subsequently, the GeneMANIA website was used to predict and construct the interaction network of the 6 key genes and their functionally similar genes (Fig. 12C). Different colors of connecting lines represent the co-expression, shared protein domains, and other information between them. The network includes 6 key genes and 20 functionally similar proteins.

PPI network of key genes. (A) PPI network of key genes calculated by the STRING database. (B) PPI network of these 6 key genes drawn using Cytoscape software. (C) Interaction network of key genes and their functionally similar genes predicted by the GeneMANIA website. The circles in the figure represent the key genes and functionally similar genes, and the colors of the connecting lines represent the functions that connect them. PPI, Protein-protein Interaction.
Construction of regulatory networks
First, the ChIPBase and hTFtarget databases were used to obtain TFs that bind to the 6 key genes, construct an mRNA-TF regulatory network, and visualize it using the Cytoscape software (Fig. 13A). The network includes 6 key genes and 49 TFs. Detailed information was in Supplementary Table S3. Subsequently, the StarBase database was utilized to identify miRNAs related to these key genes, construct an mRNA-miRNA regulatory network, and visualize it using the Cytoscape software (Fig. 13B), involving 3 key genes and 32 miRNAs. Specific information is shown in Supplementary Table S4. Finally, the StarBase database was also used to predict RBPs related to these key genes, construct an mRNA-RBP regulatory network, and visualize it using the Cytoscape software (Fig. 13C), containing 6 key genes and 116 RBPs. Detailed information is shown in Supplementary Table S5.
Finally, the CTD database was utilized to identify potential drugs or molecular compounds associated with the 6 essential genes. Cytoscape was employed to construct and visualize an mRNA-drug regulatory network (Fig. 13D), which includes 6 essential genes and 20 drugs or molecular compounds. Specific information can be found in Supplementary Table S6.

Regulatory network of key genes. (A) mRNA-TF regulatory network of key genes. (B) mRNA-miRNA regulatory network of key genes. (C) The mRNA-RBP regulatory network of key genes. (D) Key genes (mRNA-Drug Regulatory Network). TF, Transcription Factor; RBP, RNA-Binding Protein. mRNA in purple, TF in blue, miRNA in red, RBP in yellow, and Drug in green.
Discussion
IDD is a major global health problem associated with severe pain and disability, affecting hundreds of millions of people worldwide51. Existing treatment methods, such as surgery and medication, have limitations and often require a balance between treatment effects and side effects52. Therefore, in-depth research on the molecular mechanisms of IDD, especially gene expression changes in the glycosylation process, is crucial for developing new diagnostic and therapeutic strategies. This study conducted bioinformatic analysis of gene expression in IDD based on datasets downloaded from public databases.
Hypoxia is a typical feature of the microenvironment of the NP tissue, especially when intervertebral disc degeneration occurs. Due to annular rupture, reduced blood vessels, and increased metabolic demands of nucleus pulposus cells, the local partial pressure of oxygen drops sharply53. The results of GSEA suggest that the integrated dataset significantly enriches gene sets such as Adaptation to Hypoxia (down) and Hypoxia (down), indicating that the hypoxia adaptation ability of NP tissue is significantly impaired under the state of IDD. GSEA analysis shows that the differentially expressed genes in the integrated dataset are significantly enriched in the Apoptosis By Serum Deprivation pathway (up), suggesting that the apoptotic pathway is activated in IDD, which is highly consistent with the pathological features of NP tissue degeneration. As IDD progresses, the nutrient channels of the intervertebral disc calcify, and NP cells face a lack of nutrients such as glucose and amino acids, thereby initiating the apoptotic program54. GSEA analysis shows that the differentially expressed genes in the integrated dataset are significantly enriched in the Integrated TGF-β EMT pathway (up), suggesting that the TGF-β induced EMT- related pathway is activated in IDD. When IDD occurs, the TGF-β signaling pathway may drive fibrosis and NP dehydration by inducing an imbalance in the synthesis and degradation of ECM components55.
The diagnostic model constructed in this study (Fig. 7D, E) included 7 glycosylation-related key genes: MAN2B2, IGFBP3, MAN1A1, CHI3L1, PLOD2, RAPGEF5, and GLA. According to the results of differential expression gene analysis ((Fig. 3D, E), diagnostic model verification results ((Fig. 8F), and glycosylation score results (Fig. 10B, C), MAN2B2 and RAPGEF5 showed expression patterns suggestive of a protective association with IDD, whereas IGFBP3, GLA, MAN1A1, PLOD2, and CHI3L1 were correlated with IDD in this dataset. These associations require experimental validation to confirm any functional roles. Among them, MAN2B2 has the highest diagnostic efficiency for IDD diagnosis model. And we found that MAN2B2, IGFBP3, PLOD2, CHI3L1 and other genes have been confirmed by relevant studies, with high accuracy and specificity. Eva Morava and Xue Zhang56,57 reported two cases of patients with MAN2B2 mutation defect, which showed serious arthritis, malformation and immune deficiency, etc. After transduction of wild-type MAN2B2, the patients’ related symptoms were relieved. Studies of Grad et al.58 have shown that IGFBP3 can affect the dynamic balance of matrix synthesis and degradation by regulating IGF-1 activity, and the polymorphism of IGFBP3 gene is closely related to lumbar disc degeneration. Levi59 showed that PLOD2 encodes a collagen lysine hydroxylase, which is highly expressed after tissue injury and can regulate extracellular matrix remodeling by affecting collagen fiber crosslinking. Huan Wang60 showed that CHI3L1 secreted by M2a macrophages promoted the imbalance of extracellular matrix metabolism by activating IL-13Rα2/MAPK pathway, thus promoting IDD. Other genes such as RAPGEF5, GLA, MAN1A1, etc., have not been reported to be related to IDD at present, and their specific functions need to be further explored. The potential roles of these genes in IDD remain to be clarified and warrant further investigation through experimental studies.
Immune cell infiltration of IDD is another highlight of this study. A large number of studies have shown that immune cell infiltration and inflammatory response are important factors leading to IDD61,62. We used CIBERSORT algorithm to identify multiple invasive immune cell subpopulations in degenerative tissues and analyze the correlation between key genes and immune cells. Subsequently, we further analyzed the action mechanism of immune cells on IDD (Figs. 9C, 10E and 11F) and found that T cells CD4 memory resting, Neutrophils and B cells memory belonged to protective immune cells. Macrophages M0, CD56dim natural killer cell, T cells CD8 and MDSC belong to pathogenic immune cells. Eosinophils, T cells regulatory, Dendritic cells activated, and T cells follicular helper have conflicting effects on IDD. The results in Fig. 9 indicate that follicular helper T cells exhibit the highest correlation coefficient with activated dendritic cells (r > 0.5, p < 0.001). Meanwhile, there is a strong negative correlation between activated NK cells and mast cells (r < -0.5, p < 0.001). These closely associated immune cells might reflect a specific immune regulatory network within the microenvironment of IDD. CD4 memory resting T cells are moderately positively correlated with regulatory Tregs. This finding suggests that in the context of IDD, resting T cells may be activated and differentiate into Tregs, thereby participating in the suppression of inflammation. This interplay embodies the dynamic equilibrium of immune regulation. A weak positive correlation exists between eosinophils and mast cells, which could be attributed to their collaborative secretion of anti-inflammatory cytokines such as IL − 4/IL − 13. This interaction may represent an anti-inflammatory compensatory mechanism in IDD. This is consistent with the negative correlation between eosinophils and MAN2B2 (a protective gene) observed in “Immune infiltration analysis between high and low glycosylation score groups”, further corroborating the protective role of eosinophils in IDD.
Ming-Xiang Zou63 conducted single-cell RNA sequencing of intervertebral discs in IDD, suggesting that Neutrophils interfered with nucleus pulposus cells to promote the progression of IDD. In addition, a bioinformatics study64 found that imbalances in Neutrophils and γδT cells were significantly associated with IDD progression. However, this study suggests that MAN1A1 gene may inhibit IDD by regulating Neutrophils. Zhengxu Ye65 suggested that M1 polarization of macrophages could accelerate disc degradation and promote IDD. Our analysis suggests a possible association between M0-to-M1 macrophage polarization and IDD, and genes such as CHI3L1, MAN1A1, and PLOD2 may be linked to this process. However, these hypotheses require experimental confirmation. MDSC expression is elevated in inflammatory and chronic diseases. This study suggests that CHI3L1 and PLOD2 genes may promote IDD by regulating MDSC. Juan Du66 showed that circulating MDSCs were significantly positively correlated with the severity of clinicopathological stages of LDH. Yang Sun67 have shown that eosinophils regulate the polarization of macrophages by secreting cytokines such as IL-4 and IL-13, and have anti-inflammatory effects. Therefore, it is speculated that eosinophils may play a protective role in IDD through this mechanism, and MAN2B2, GLA, CHI3L1 and other genes may participate in this process by regulating eosinophils. For other immune cells, such as T cells CD4 memory resting, B cells memory, T cells CD8, etc., no studies have been found on IDD, and its mechanism is still unclear. In summary, this study found that a variety of immune cells have different effects on IDD, and some immune cells have contradictory effects on IDD, suggesting the importance and complexity of immune cell infiltration in IDD, which needs to be further studied.
On the other hand, this study used the ssGSEA algorithm to perform clustering analysis on IDD samples based on the infiltration levels of 28 immune cell types and divided the samples into two immune subtypes. By comparing the expression differences of key genes and immune cell infiltration characteristics between the two subtypes, it was found that MAN1A1 and IGFBP3 were highly expressed in subtype 2, and the two subtypes had significant differences in the infiltration levels of 12 immune cell types, such as activated CD4 T cells, activated dendritic cells, and effector memory CD8 T cells. Studies have shown that immune cell infiltration patterns in the IDD process have stage specificity, with macrophage infiltration predominating in the early stage, while T lymphocyte and dendritic cell infiltration increases in the middle and late stages68. The results of the clustering analysis in this study support this view, suggesting that changes in the immune microenvironment play an important role in the formation of IDD heterogeneity, and different immune subtypes may correspond to different stages or severities of the disease. Therefore, immune phenotypes may become a new type of molecular marker for judging the degree of degeneration and guiding the selection of treatment plans.
In terms of revealing the biological functions of key genes, we found that multiple TFs, such as CEBPB and FOXA1, may be involved in the disease process by regulating the expression of IGFBP3, PLOD2, and other genes. In addition, miRNAs such as miR-19a-3p and miR-96-5p may also affect intervertebral disc homeostasis by targeting IGFBP3, PLOD2, and other genes. Studies have found that circARL15 plays a key role in IDD by regulating DISC1 expression through miR-431-5p69. This study further analyzed the regulatory role of key miRNAs in IDD and their association with previous research. By reviewing existing literature on the identified miRNAs, it was found that some miRNAs (such as miR-19a-3p, miR-96-5p, etc.) have been previously confirmed to be closely related to the occurrence and progression of IDD. miR-19a-3p can influence the survival status of disc cells by regulating apoptosis and inflammatory responses, while miR-96-5p plays a significant role in extracellular matrix metabolism and tissue repair processes70. The miRNAs predicted in this study are highly consistent with these known functions, further validating the reliability and biological significance of the bioinformatic screening results. However, the roles of some miRNAs also exhibit variations across different studies, potentially influenced by factors such as tissue type, sample source, or analysis strategies. Therefore, in this study, we supplemented the regulatory networks and potential target genes associated with IDD for these miRNAs and explored the functions of newly discovered miRNAs and their possible involvement in IDD pathogenesis. Overall, the miRNA network identified in this study provides a theoretical foundation and data support for understanding IDD’s molecular regulatory mechanisms and identifying novel diagnostic and therapeutic targets. Common environmental pollutants such as bisphenol A and tetrachlorodibenzodioxin can act on multiple key genes. Epidemiological studies have shown that exposure to environmental toxins such as tobacco and dioxins is a risk factor for IDD71. This suggests that exogenous chemical substances may promote the disease process by interfering with the expression of key genes, and the specific mechanisms still require more experimental research. In summary, this study explored the molecular mechanisms of IDD from the perspective of glycosylation abnormalities using bioinformatic methods, which may provide new methods and strategies for improving the prognosis of IDD patients.
Limitations of the study
This study still has certain limitations. Firstly, although the sample size was expanded by integrating multiple public databases, the combined dataset only included 21 IDD samples and 13 control samples, which is a limited sample size that may not fully represent the gene expression characteristics of IDD patients, thereby affecting the generalizability and reliability of the results. Although current research has mitigated the risk of overfitting by removing batch effects, reducing gene redundancy, and conducting cross-validation, future studies still need to incorporate external datasets for validation to further enhance the robustness and reliability of the results. Secondly, due to research constraints, it was not possible to independently collect clinical specimens or conduct related experimental validations, and all analyses were based on data from public databases. Consequently, there is a lack of validation at the protein level and functional experiments for key genes, preventing further elucidation of their specific mechanisms of action. Additionally, the limitations in sample size and experimental design made it impossible to systematically compare the expression differences of key genes in different degenerative tissue regions (such as the nucleus pulposus and annulus fibrosus). This study primarily focused on gene expression analysis of nucleus pulposus tissue, whereas the common degenerative phenotypes observed in clinical imaging often originate from annulus fibrosus lesions. In future research, we plan to expand the collection of clinical samples further, incorporating multicenter and multi-type tissue samples, and conduct protein-level and functional experimental validations to confirm and extend the conclusions of this study more comprehensively.
Data availability
Publicly available datasets (GSE34095, GSE70362, GSE147383) were analyzed in this study. All the datasets were obtained from the GEO (https://www.ncbi.nlm.nih.gov/geo/) database.
References
Hernandez, P. A., Jacobsen, T. D. & Chahine, N. O. Actomyosin contractility confers mechanoprotection against TNFalpha-induced disruption of the intervertebral disc. Sci. Adv. 6 (34), a2368 (2020).
Ravindra, V. M. et al. Degenerative lumbar spine disease: estimating global incidence and worldwide Volume. Global Spine J. 8 (8), 784–794 (2018).
Karasik, D. et al. Abdominal aortic calcification and exostoses at the hand and lumbar spine: the framingham study. Calcif Tissue Int. 78 (1), 1–8 (2006).
Cazzanelli, P. & Wuertz-kozak, K. MicroRNAs in intervertebral disc degeneration, apoptosis, inflammation, and mechanobiology. Int. J. Mol. Sci. 21(10), 3601 (2020).
Eichler J. Protein glycosylation. Curr. Biol. 29 (7), R229–R231 (2019).
Peixoto, A. et al. Protein glycosylation and tumor microenvironment alterations driving cancer Hallmarks. Front. Oncol. 9, 380 (2019).
Schedin-weiss, S., Winblad, B. & Tjernberg, L. O. The role of protein glycosylation in alzheimer disease. FEBS J. 281 (1), 46–62 (2014).
Pabst, M. et al. IL-1beta and TNF-alpha alter the glycophenotype of primary human chondrocytes in vitro. Carbohydr. Res. 345 (10), 1389–1393 (2010).
Zhang, Y. et al. MAPK8 and CAPN1 as potential biomarkers of intervertebral disc degeneration overlapping immune infiltration, autophagy, and ceRNA. Front. Immunol. 14, 1188774 (2023).
Huang, H. et al. Vitamin D retards intervertebral disc degeneration through inactivation of the NF-kappaB pathway in mice. Am. J. Transl Res. 11 (4), 2496–2506 (2019).
Zhao, R. et al. Nucleus pulposus cell senescence is regulated by substrate stiffness and is alleviated by LOX possibly through the integrin beta1-p38 MAPK signaling pathway. Exp. Cell. Res. 417 (2), 113230 (2022).
Wu, Z. L. et al. SKI knockdown suppresses apoptosis and extracellular matrix degradation of nucleus pulposus cells via Inhibition of the Wnt/beta-catenin pathway and ameliorates disc degeneration. Apoptosis 27 (1–2), 133–148 (2022).
Zhao, Y. et al. Conjoint research of WGCNA, single-cell transcriptome and structural biology reveals the potential targets of IDD development and treatment and JAK3 involvement. Aging (Albany NY). 15 (24), 14764–14790 (2023).
Hu, S. et al. Analysis of key genes and pathways associated with the pathogenesis of intervertebral disc degeneration. J. Orthop. Surg. Res. 15 (1), 371 (2020).
Zhu, Z. et al. Integrative bioinformatics analysis revealed mitochondrial Dysfunction-Related genes underlying intervertebral disc Degeneration. Oxid. Med. Cell. Longev. 1372483. https://doi.org/10.1155/2022/1372483 (2022).
Ikuno, A. et al. Genome-wide analysis of DNA methylation profile identifies differentially methylated loci associated with human intervertebral disc degeneration. PLoS One. 14 (9), e222188 (2019).
Liu, X. W. et al. Role of ferroptosis and immune infiltration in intervertebral disc degeneration: novel insights from bioinformatics analyses. Front. Cell. Dev. Biol. 11, 1170758 (2023).
Yan, Q. et al. Bioinformatics-Based research on key genes and pathways of intervertebral disc Degeneration. Cartilage 13 (2_suppl), 582S–591S (2021).
Zhang, Z. et al. GREM1, LRPPRC and SLC39A4 as potential biomarkers of intervertebral disc degeneration: a bioinformatics analysis based on multiple microarray and single-cell sequencing data. BMC Musculoskelet. Disord. 24 (1), 729 (2023).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 41 (Database issue), D991–D995 (2013).
Davis, S. & Meltzer, P. S. GEOquery: a bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics 23 (14), 1846–1847 (2007).
Stelzer, G. et al. The genecards suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinf. 54, 1–30 (2016).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27 (12), 1739–1740 (2011).
Chen, L. & Ling, Y. Yang, H. Comprehensive analysis of the potential prognostic value of 11 glycosylation-related genes in head and neck squamous cell carcinoma and their correlation with PD-L1 expression and immune infiltration. J. Oncol. 2022, 2786680. (2022).
Ben S. K. & Ben, A. A. Principal component analysis (PCA). Tunis Med. 99 (4), 383–389 (2021).
Zhang, H. & Meltzer, P. RCircos: an R package for circos 2D track plots. BMC Bioinform. 14, 244 (2013).
Mi, H. et al. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47 (D1), D419–D426 (2019).
Kanehisa, M. KEGG: Kyoto encyclopedia of genes and Genomes. Nucleic Acids Res. 28 (1), 27–30 (2000).
Kanehisa M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28 (11), 1947–1951 (2019).
Kanehisa, M. et al. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51 (D1), D587–D592 (2023).
Yu, G. et al. ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16 (5), 284–287 (2012).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 102 (43), 15545–15550 (2005).
Liu, W. et al. Weighted gene co-expression network analysis in biomedicine research . Sheng Wu Gong. Cheng Xue Bao. 33 (11), 1791–1801 (2017).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Sanz, H. et al. SVM-RFE: selection and visualization of the most relevant features through non-linear kernels. BMC Bioinform. 19 (1), 432 (2018).
Engebretsen, S. & Bohlin, J. Statistical predictions with glmnet. Clin. Epigenetics. 11 (1), 123 (2019).
Wu, J. et al. A nomogram for predicting overall survival in patients with low-grade endometrial stromal sarcoma: A population-based analysis. Cancer Commun. (Lond). 40 (7), 301–312 (2020).
Van Calsterb, B. et al. Reporting and interpreting decision curve analysis: a guide for Investigators. Eur. Urol. 74 (6), 796–804 (2018).
Newman, A. M., et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 12 (5), 453–457 (2015).
Xiao, B. et al. Identification and verification of Immune-Related gene prognostic signature based on SsGSEA for Osteosarcoma. Front. Oncol. 10, 607622 (2020).
Lock, E. F. & Dunson, D. B. Bayesian consensus clustering. Bioinformatics 29 (20), 2610–2616 (2013).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26 (12), 1572–1573 (2010).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47 (D1), D607–D613 (2019).
Franz, M. et al. GeneMANIA update 2018. Nucleic Acids Res. 46 (W1), W60–W64 (2018).
Zhou, K. R. et al. ChIPBase v2.0: decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res. 45 (D1), D43–D50 (2017).
Zhang, Q. et al. hTFtarget: a comprehensive database for regulations of human transcription factors and their Targets. Genomics Proteom. Bioinf. 18 (2), 120–128 (2020).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13 (11), 2498–2504 (2003).
Li, J. H. et al. StarBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42(Database issue), D92–D97. https://doi.org/10.1093/nar/gkt1248 (2014).
Singh, A. RNA-binding protein kinetics. Nat. Methods. 18 (4), 335 (2021).
Grondin, C. J.,et al. Predicting molecular mechanisms, pathways, and health outcomes induced by Juul e-cigarette aerosol chemicals using the comparative toxicogenomics Database. Curr. Res. Toxicol. 2, 272–281 (2021).
Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet 392 (10159), 1789–1858 (2018).
Taso, M. et al. A randomised controlled trial comparing the effectiveness of surgical and nonsurgical treatment for cervical radiculopathy. BMC Musculoskelet. Disord. 21 (1), 171 (2020).
Kim, H. J. et al. Hypoxia Helps Maintain Nucleus Pulposus Homeostasis by Balancing Autophagy and Apoptosis. Oxid Med Cell Longev, 2020: 5915481. (2020).
Yang, F. et al. Regulated cell death: implications for intervertebral disc degeneration and therapy. J. Orthop. Translat. 37, 163–172 (2022).
Chen, S. et al. TGF-β signaling in intervertebral disc health and disease. Osteoarthr. Cartil. 27 (8), 1109–1117 (2019).
Verheijen, J. et al. Defining a new immune deficiency syndrome: MAN2B2-CDG. J. Allergy Clin. Immunol. 145 (3), 1008–1011 (2020).
Fan, S. et al. Congenital disorders of glycosylation with multiorgan disruption and immune dysregulation caused by compound heterozygous variants in MAN2B2. Mol. Genet. Genomic Med. 12 (4), e2422 (2024).
Kazezian, Z. & Gawri, R. Gene expression profiling identifies interferon signalling molecules and IGFBP3 in human degenerative annulus Fibrosus. Sci. Rep. 5, 15662 (2015).
Kang, H. et al. The HIF-1alpha/PLOD2 axis integrates extracellular matrix organization and cell metabolism leading to aberrant musculoskeletal repair. Bone Res. 12 (1), 17 (2024).
Li, L. et al. M2a Macrophage-Secreted CHI3L1 promotes extracellular matrix metabolic imbalances via activation of IL-13Rα2/MAPK pathway in rat intervertebral disc Degeneration. Front. Immunol. 12, 666361 (2021).
Nakazawa, K. R. et al. Accumulation and localization of macrophage phenotypes with human intervertebral disc degeneration. Spine J. 18 (2), 343–356 (2018).
Risbud, M. V. & Shapiro, I. M. Role of cytokines in intervertebral disc degeneration: pain and disc content. Nat. Rev. Rheumatol. 10 (1), 44–56 (2014).
Zhang, T. L. et al. Single-cell RNA sequencing reveals the MIF/ACKR3 receptor-ligand interaction between neutrophils and nucleus pulposus cells in intervertebral disc degeneration. Transl Res. 272, 1–18 (2024).
Li, W. et al. The roles of blood Lipid-Metabolism genes in immune infiltration could promote the development of IDD. Front. Cell. Dev. Biol. 10, 844395 (2022).
Zhao, X. et al. Degenerated nucleus pulposus cells derived exosome carrying miR-27a-3p aggravates intervertebral disc degeneration by inducing M1 polarization of macrophages. J. Nanobiotechnol. 21 (1), 317 (2023).
Zhou, H. et al. Increased levels of Circulating granulocytic myeloid-derived suppressor cells in lumbar disc herniation. Exp. Ther. Med. 26 (2), 367 (2023).
Jian, Y. & Zhou, X. Crosstalk between macrophages and cardiac cells after myocardial infarction. Cell. Commun. Signal. 21 (1), 109 (2023).
Molinos, M. et al. Inflammation in intervertebral disc degeneration and regeneration. J. R Soc. Interface. 12 (104), 20141191 (2015).
Wang, H. et al. circARL15 plays a critical role in intervertebral disc degeneration by modulating miR-431-5p/DISC1. Front. Genet. 12, 669598 (2021).
Li, X., Hou, Q., Yuan, W. & Zhan, X. Inhibition of miR-96-5p alleviates intervertebral disc degeneration by regulating the peroxisome proliferator-activated receptor γ/nuclear factor-kappaB pathway. J. Orthop. Surg. Res. 18 (1), 916 (2023).
Elmasry, S. & de Asfour, S. Effects of tobacco smoking on the degeneration of the intervertebral disc: a finite element study. PLoS One. 10 (8), e136137 (2015).
Acknowledgements
We thank all those who have contributed sequences to NCBI databases.
Funding
This work was supported by the National Natural Science Foundation of China (82074529), Li Yikai Guangdong Province Famous Traditional Chinese Medicine Inheritance Studio Construction Project (Guangdong Traditional Chinese Medicine Office Letter 2023_108), Sanming Project of Medicine in Shenzhen (SZZYSM202108013), 2026 Zhejiang Provincial Traditional Chinese Medicine Science and Technology Plan (2026ZL0418), the Research Project of Zhejiang Chinese Medical University (2025RCZXZK76), and Science and Technology Projects in Guangzhou (2024A04J5180).
Author information
Authors and Affiliations
Contributions
Gaofeng Liu: writing – original draft, visualization, investigation, conceptualization. Qingguang Qin: visualization, software, methodology, funding acquisition. Weixing Zhong: visualization, software, data curation. Hongwen Liu: visualization, software. Yin Xie: visualization, software. Xiaoli Huang: visualization, software. Guangjie Liu: writing – review & editing, validation, supervision, methodology, formal analysis. Yikai Li: writing – review & editing, supervision, resources, project administration, funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, G., Qin, Q., Zhong, W. et al. Bioinformatic analysis of glycosylation-related genes in intervertebral disc degeneration and their roles in immune infiltration and diagnostic models. Sci Rep 15, 44067 (2025). https://doi.org/10.1038/s41598-025-27197-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-27197-1
