
Abstract
PROteolysis-Targeting Chimeras (PROTACs) are an emerging class of molecules capable of inducing a forced approximation between a protein of interest (POI) and an E3 ligase enzyme (e.g., Cereblon), leading to the degradation of the POI by the cell’s own machinery. Although in early stages of development, PROTACs’ unique mechanisms of action offer a novel therapeutic strategy, which has attracted growing interest worldwide. Cereblon-based PROTACs are the most studied class of PROTACs and have been actively researched in recent years for the treatment of different diseases, from cancer to neurological disorders, with some of them already in clinical trials. In this review, we provide a comprehensive and critical analysis covering the recent advances, potential challenges and future prospects regarding the design and synthesis, as well as pre- and clinical evaluation of cereblon-based PROTACs. By integrating insights from drug discovery and development, a broad yet in-depth discussion is given to guide future research on cereblon-based PROTACs.

Similar content being viewed by others
Introduction
Involved in almost all biological and pathological mechanisms, whether inside or outside cells, proteins are one of the most important building blocks of life1. Throughout their entire life cycle, proteins are subject to tight regulation at the pre-, co- and post-translational levels, which is critical for the maintenance of proteostasis2,3. The coordinated and precise interaction between the ubiquitin-proteasome system (UPS), the protein chaperone system, and the autophagy-lysosome system (ALS) results in a dynamic endogenous protein quality control system that is essential to ensure the protein homeostasis4,5,6. Focusing on the UPS, this is the main proteolysis pathway for short-lived, damaged, oxidized, misfolded, or abnormal proteins7. It is also involved in the normal turnover process of proteins that have fulfilled their functions8,9. Simplistically, the UPS can be divided into two subsystems: the ubiquitination system, which is responsible for the addition of ubiquitin moieties to target proteins, and the proteasome system, which is responsible for the recognition and degradation of poly-ubiquitinated proteins10,11. Ubiquitin-mediated protein degradation by the UPS was discovered in the 1980s, and begins with the poly-ubiquitination of the target, that is, with the addition to the protein to be degraded of several ubiquitin units - a 76-aminoacid regulatory protein of globular conformation12,13. However, the ubiquitination state of a protein also defines, for example, its cellular location, or its recognition by signaling or regulatory complexes14,15. From the sequential activity of ubiquitin-activating enzymes (E1), ubiquitin–conjugating enzymes (E2), and ubiquitin protein ligases (E3), ubiquitin is catalytically added to the target in an ATP-dependent process16. Target specificity is ensured by direct binding between the E3 ligase, which recognizes the substrate and transfers the ubiquitin units to its surface17. The ATP-dependent proteolytic complex, called the 26S proteasome, then recognizes the polyubiquitin protein and degrades it into small peptide fragments18.
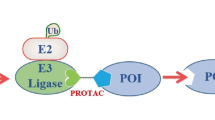
In 2001, the drug discovery paradigm was completely revolutionized by the pioneering work of Crews and Deshaies, who published the first compound capable of directing the UPS against a protein of interest (POI), thereby forcing its non-natural degradation (Fig. 1)19. This successful proof-of-concept study opened the doors to the PROteolysis-TArgeting Chimeras (PROTACs) development. PROTACs are heterobifunctional molecules containing a pharmacophoric moiety capable of binding to the POI (target ligand moiety (TLM)) at one end and an E3 ligase binding moiety (ELM) capable of recruiting an E3 binding enzyme from the UPS at the other end20. The connection between these two ligands (TLM and ELM) is established by a chemical linker21. Mechanistically, once inside the cell, PROTAC forms a stable ternary complex resulting from its simultaneous binding to the POI and a specific E3 ligase22. By forcing proximity between the target protein and the E3 ligase, PROTAC induces proximity-driven polyubiquitination of the POI, i.e., the proximity forced by the PROTAC results in the transfer of the ubiquitin units from the E3 ligase to the POI23. After polyubiquitination, the target protein dissociates from PROTAC and follows the normal UPS pathway, where it is recognized by the 26S proteasome and degraded into small peptide fragments24. Afterwards, the PROTAC is free to form a new stable ternary complex with another POI, thus exhibiting an advantageous catalytic mechanism of action (event-driven mechanism)25. Twenty-five years after the publication of the first PROTAC, these are included within the Targeted Protein Degradation (TPD) field, which encompasses all innovative strategies aimed at chemically-induced protein degradation, known generically as degraders26. However, unquestionably PROTACs are the most popular within the TPD field27.

In the Ubiquitin-Proteasome System (UPS), the process of ubiquitination begins when the ubiquitin protein (Ub) is activated by the ubiquitin-activating E1 enzyme. The activated Ub is then transferred to the ubiquitin-conjugating enzyme E2. Afterwards, the E3 ubiquitin ligase connects both to the target protein and to the E2-Ub complex, enabling the attachment of ubiquitin molecules to the target. Once tagged with a polyubiquitin chain, the target protein is recognized and degraded by the 26S proteasome. PROTACs are heterobifunctional molecules containing a target protein ligand at one end, and an E3 ligase ligand capable of recruiting an E3 ligase enzyme from the UPS to the other end, with a linker establishing the connection between the two ligands. Within the cell, PROTAC promotes the simultaneous and artificial binding of the target protein to an E3 ligase, forming a stable ternary complex. This leads to the target poly-ubiquitination and subsequent non-natural degradation by the UPS.
The PROTACs boom was sparked by several causes. Firstly, due to its revolutionary mechanism of action, in which the suppression of the biological effect is obtained through the degradation of the therapeutic target and protein aggregates, using the cell’s own machinery20. Secondly, due to the ability to suppress all functions of a given protein, as well as “undruggable proteins” (e.g., transcription factors or scaffold proteins), since PROTAC does not need to bind to a specific site on the target protein22. Third, due to preclinical and clinical outcomes that have demonstrated that PROTACs have the potential to overcome many of the challenges observed with the use of small molecular inhibitors (SMIs), such as the development of resistance or serious side effects27. The consolidation of the therapeutic potential of PROTACs has led to an intensification of the race to discover better PROTACs28.
In general, PROTACs are classified according to the E3 ligase recruited. To date, of the more than 600 different E3 ligases identified in human cells, the vast majority of reported PROTACs, including those in clinical studies, recruit the cereblon E3 ligase29. The second most commonly recruited E3 ligase by PROTACs is the Von Hippel-Lindau (VHL) E3 ligase29. To a lesser extent, PROTACs also recruit the inhibitor of apoptosis protein (IAP) and the mouse double minute ligases (MDM2)29. Recently, the activity of new PROTACs that recruit other E3 ligases, such as the Kelch-like ECH-associated protein 1 (KEAP1) E3 ligase, the DDB1-CUL4A-associated factor 16 (DCAF16) E3 ligase and the RING finger protein 114 (RNF114) E3 ligase, has been investigated30,31,32. There are numerous articles demonstrating that cereblon-based PROTACs can rapidly, potently and highly selectively degrade, both in vitro and in vivo, a huge number of different proteins associated with the development of various diseases, including cancer, neurodegenerative diseases, immune diseases, cardiovascular diseases and viral infections33,34.
Undoubtedly, cereblon-based PROTACs are frontrunners in the PROTAC field, and have aroused global interest from the pharmaceutical industry and academia35. Therefore, this comprehensive review aims to provide an up-to-date critical overview on cereblon-based PROTACs, covering the main aspects related to the chemical synthesis of cereblon ligands derivatives, the chemical ligation of these derivatives to the linkers, and the construction of the final cereblon PROTAC. Furthermore, relevant and recent examples of novel cereblon-based PROTACs will be presented and the main results obtained from their biological evaluation will be analyzed. In this review, potential challenges and future perspectives of PROTACs that recruit the cereblon E3 ligase will also be discussed.
Cereblon—structure and function
In 2004, Joseph J. Higgins described the cereblon protein for the first time, after mapping the deletion and mutation of its gene in a patient with autosomal recessive non-syndromic mental retardation36. Cereblon designation derives from its high expression in the cerebellum, its importance in mental development, as well as its highly conserved ATP-dependent Lon domain37. Among the various domains of cereblon, the drug-binding region called cereblon domain stands out, which is responsible for recognizing and binding to endogenous target proteins, as well as specific immunomodulatory imide drugs (IMiDs), with emphasis on thalidomide, lenalidomide and pomalidomide4.
Cereblon encodes a 442 amino acid protein, with a molecular weight of 51 kDa, which is widely expressed in all tissues4,37,38. Cereblon functions as a substrate specificity receptor/substrate recognition protein within the Cullin 4-really interesting new gene (RING) E3 ubiquitin ligase CRL4CRBN complex39. With approximately 400 different members, the CRL4 subfamily is the largest group of cullin RING E3 ligases38. The CRL4CRBN complex is composed of cereblon, which interacts with damaged DNA-binding protein 1 (DDB1), Cullin4 (Cul4A/4B), and regulator of cullins 1 (RBX1)40. In this complex, the cereblon is the unit responsible for recognizing and binding to target proteins for subsequent ubiquitination, while the DDB1 protein functions as a support linking cereblon to the Cullin4A/4B proteins, which in turn serve as molecular scaffolds33,41. Although the intracellular location of Cereblon depends on the cell type in question, in general, it is distributed throughout the cell, including in the nucleus, given that the N-terminal region of cereblon can function as an important signal for nuclear localization4,37.
Over the last few decades, different targets of cereblon have been identified, concluding that the targets vary according to the tissue and cellular location of this E3 ligase42. Cereblon has gained prominence as a potential therapeutic target in several diseases since, by interacting with several proteins involved in multiple downstream signaling pathways, it ends up controlling a wide range of different physiological processes crucial for normal cellular functioning4,37. For example, although not all of its targets are known, many are involved in the regulation of ion channels, immune system, and metabolism38. Some of its targets have also been implicated in the development of many diseases, including cerebral, cardiovascular, and oncological diseases38. Recently, the ubiquitination-independent functions of cereblon have been studied, which include its function as a transcription repressor, chaperone protein, and scaffold protein37.
Cereblon ligands
Molecular mechanistic studies with the controversial drug thalidomide led to the discovery of cereblon’s ligands43. Developed in the 1950s by the German pharmaceutical company Chemie Grünenthal, thalidomide was initially administered as a sedative-hypnotic drug, widely prescribed at the time to treat morning sickness in pregnant women21,37. However, a few years later, it was withdrawn from the market due to its severe teratogenicity (>10,000 cases of phocomelia birth defects worldwide) in what became known as the thalidomide tragedy, one of the most devastating in the pharmaceutical industry history44,45.
After a few decades, studies showed that thalidomide and its structural analogs have anti-inflammatory, anti-angiogenic and anti-myeloma properties, thus leading to their designation as IMiDs37,43. However, it was only in 2010 that Ito’s group demonstrated that thalidomide binds to cereblon E3 ligase46. Additional studies have demonstrated that the biologicals effects of thalidomide and IMiDs result from their binding to cereblon, which culminates in the ubiquitination and degradation of neo-substrates47. For example, lenalidomide and pomalidomide lead to the degradation of the neo-substrates Ikaros (family zinc finger protein-1, IKZF1) and Aiolos (family zinc finger protein-3, IKZF3) transcriptional factors, thereby exhibiting an anti-myeloma activity48,49.
Structurally, the cereblon E3 ligase ligands have a phthalimide unit linked to a glutarimide unit moiety (Fig. 2)50. Studies by Fischer et al. to characterize the structure of the cereblon complex and its binding to IMiDs showed that thalidomide, pomalidomide and lenalidomide bind very similarly to cereblon protein51. More specifically, in the phthalimide part, the phenyl group is solvent-exposed, while one of the carbonyl groups forms the water-mediated hydrogen bond with His35921. The glutarimide moiety occupies a conserved hydrophobic pocket on the C-terminal domain of cereblon, also known as the cereblon domain, with the carbonyl groups and intervening amide interacting with His380 and Trp382 via hydrogen bonding51. The remaining carbon atoms of the glutarimide ring interact with the amino acids Trp382, Trp388, Trp402, and Phe404 through van der Waals bonds51. The structural analysis of thalidomide and its structural analogs pomalidomide and lenalidomide reveals the presence of a chiral center, which is known to influence the binding affinity to cereblon protein. However, as these compounds racemize under physiological conditions, they are administered as racemic mixtures52. In the following schemes, stereochemical information is provided wherever possible. If this does not occur, it is either because the compounds exist in the form of an isomeric mixture, or because such information is not reported in the literature.

A Chemical structure of immunomodulatory drugs (IMiDs): thalidomide, pomalidomide and lenalidomide. B Interactions between aminoacids residues of cereblon E3 ligase and thalidomide.
Following the discovery of the binding capacity of IMiDs drugs with cereblon E3 ligase, the focus in this area over the last few years has been on the synthesis and study of new structural analogs of IMiDs with the aim of obtaining new and better building blocks for the construction of cereblon-recruiting PROTACs43,53.
Cereblon-based PROTACs—from synthesis to biological evaluation
At the moment, cereblon-based PROTACs are the frontrunners in the PROTACs field. PROTACs recruiting the cereblon E3 ligase have successfully induced the forced degradation of more than 60 different proteins, associated with the development of a wide range of disorders, from cancer to neurological, immunological and viral diseases, with successful examples already in clinical trials54,55,56,57,58.
The study of thalidomide chemistry led to the differentiated functionalization of the phthalimide ring, allowing the production of different cereblon ligand libraries for incorporation into the construction of novel cereblon-based PROTACs (Table 1)59.
To date, cereblon-based PROTACs are the most commonly reported in both the scientific literature and patents, due to the advantages presented by the incorporation of cereblon ligands, compared to the incorporation of ligands of other E3 ligases. Therefore, the success of cereblon-based PROTACs is due to a set of key factors: Firstly, the most commonly used cereblon ligands are IMiDs, as they exhibit highly specific, strong and biophysically validated E3 ligase binding affinity43; Secondly, cereblon ligands have desirable physicochemical properties, including low molecular weight, lipophilicity, solubility, chemical and metabolic stability and the absence of pan-assay interference compound (PAINS) warnings60. It should be noted that these characteristics are favorable for obtaining PROTACs with satisfactory oral bioavailability; Thirdly, the cereblon ligands present a faithfully characterized structural information about their binding mode to cereblon protein51; Fourthly, cereblon ligands are versatile and easily modifiable61; Fifthly, cereblon E3 ligase is expressed in multiple tissues, enabling cereblon-based PROTACs to treat various diseases62. On the other hand, VHL ligands, although also easily modifiable, are large and lipophilic molecules, which can lead to solubility and permeability problems in PROTACs63. Currently available MDM2 ligands may undesirably interfere with the p53 cycle, causing serious adverse effects64. Regarding the ligands of E3 ligases less recruited by PROTACs, such as KEAP1 or DCAF, the existing ligands are still scarce, with low affinities and little studied. Therefore, the result of the desirable properties obtained with the incorporation of cereblon ligands in the construction of PROTACs, together with the excellent therapeutic results published in recent years, obtained by forced induction of the degradation of a specific POI by cereblon-based PROTACs in vitro, ex vivo and in vivo, and for different types of diseases, make this type of PROTACs the leaders in the field of TPD. Consequently, the race to discover promising PROTACs recruiting the cereblon E3 ligase has been fierce in both academia and big pharma, as reflected by the dramatic increase in the number of scientific papers and patents filed in recent years65,66.
Although a wide range of cereblon ligands that can be used to construct PROTACs are currently reported, most of cereblon-recruiting PROTACs results from attaching different types of linkers to pomalidomide, thalidomide and lenalidomide derivatives. More recently, the incorporation of 5-phthalimide substituted derivatives and novel non-phthalimide cereblon binders have been reported.
Regarding the synthetic pathway of cereblon-based PROTACs, there are conventionally three approaches to assemble the different building blocks (i.e., target ligand, linker and cereblon ligand) (Fig. 3):
-
a.
First bind the cereblon ligand to the desired linker, and then bind it to the target ligand (Approach A);
-
b.
First bind the linker to the target ligand, and then bind it to the cereblon ligand (Approach B);
-
c.
Attaching two smaller linker fragments separately to the target and cereblon ligands, then joining the two halves together (Approach C).

Three conventional approaches are used: (Approach A) coupling the cereblon ligand to the linker first, followed by attachment of the target ligand; (Approach B) coupling the linker to the target ligand first, then attaching the cereblon ligand; and (Approach C) connecting two smaller linker fragments separately to the target and cereblon ligands, followed by joining the two halves to form the complete PROTAC molecule.
In each of the above approaches, it is possible to modify the warhead (i.e., target and cereblon ligands) to facilitate or optimize linker conjugation. The choice of the best approach for the construction of cereblon-based PROTACs depends on factors such as the availability and cost of the building blocks, or the number of steps and purification processes required to obtain the final compound.
For clarity and organizational purposes, this review article categorizes cereblon-based PROTACs according to the cereblon ligand, and further subdivides them based on the type of bond used for linker attachment. In each section, a critical and up-to-date review is provided, covering not only the design and synthetic strategies of PROTACs, but also a detailed study of the chemical synthesis of key building blocks and their attachment to linkers. This review also covers important aspects related to the biological evaluation of cereblon-based PROTACs, with relevant examples published in recent years.
Pomalidomide-based PROTACs
Pomalidomide-based ligands can generally be synthesized via two different routes. One route is via condensation between a glutarimide ring unit with the 3-fluorophthalic anhydride (1), followed by a nucleophilic aromatic substitution (SNAr) reaction with a linker whose terminus is a primary or secondary amine61. Another route is via condensation of the glutarimide ring unit with the 3-nitrophthalic anhydride, with consequent reduction of the nitro group to an amine group, thus producing pomalidomide64.
In general, the current synthesis methods for pomalidomide-based PROTACs are low yielding and produce undesirable by-products61,64.
Pomalidomide-based PROTACs from 3-fluorophtalic anhydride
One of the most widely used strategies for linker attachment in PROTACs is via SNAr reactions with fluorinated derivatives of thalidomide.
When the starting compound is 4-fluorothalidomide (2), it is possible to synthesize alkylated pomalidomide derivatives (4) by SNAr reactions between 4-fluorothalidomide and linkers with primary or secondary amine end groups, in the presence of N,N-diisopropylethylamine (DIPEA) and using dimethylformamide (DMF) or dimethyl sulfoxide (DMSO) as solvent, at high temperatures (>90 °C) (Scheme 1)67,68. Although DMF is a common solvent used in this type of reaction, the high temperatures associated with the presence of tertiary amines (e.g., DIPEA) can result in thermal decomposition of the DMF into a dimethylamine by-product. In turn, dimethylamine can react with 4-fluorothalidomide and form the undesirable product 4-(dimethylamino)-thalidomide (5)69,70,71. Replacement of DMF with DMSO, dioxane or dimethylacetamide (DMA) has been reported to avoid the formation of this by-product during the linker attachment step69,72. It should further be noted that performing these reactions at higher temperatures (>120 °C) generally translates into a higher yield of the desired alkylated pomalidomide derivative71. Chen et al. similarly determined that the SNAr time can be reduced through its execution in N-methyl-2-pyrrolidone (NMP) under microwave irradiation, but the yields are not better73.

Chemical synthesis of pomalidomide-based PROTACs from 3-fluorophtalic anhydride. Reagents and conditions: a1 3-aminopiperidine-2,6-dionehydrochloride, triethylamine, THF, 80 °C; a2 3-aminopiperidine-2,6-dionehydrochloride, sodium acetate, acetic acid, 140 °C, 12 h; b 1, sodium acetate, acetic acid, reflux, 6 h; c DIPEA, DMF or DMSO, 90 °C to 130 °C, 6 to 16 h (depending on the type of linker).
To improve yields and chemoselectivity in the production of pomalidomide-based PROTACs, Derksen’s group published an interesting paper investigating the effect of solvent choice on chemical yields71. This study demonstrated that the choice of DMSO (polar aprotic solvent) as reaction solvent allows to obtain the best yields with both primary (yield > 50%) and secondary amines (yield > 90%)71. Interestingly, it was also found that, in general, with linker bearing the primary amine terminal present lower yields, when compared with the use of secondary amines under the same experimental conditions71. This may be due to the formation of the by-product from the displacement of aminoglutarimide and the lower reactivity inherent to primary amines71.
There are several examples of cereblon-based PROTACs recently reported in the literature starting from the building block 4-fluorothalidomide (Table 2).
In 2024, Zang et al. synthesized and evaluated the antitumor activity of novel PROTACs against programmed cell death ligand protein 1 (PD-L1). From the analysis of the structure-activity relationship (SAR) studies, the pomalidomide-based PROTACs showed better results than the lenalidomide-based or VHL-based PROTACs. Among the first, the best PROTACs were those incorporating a polyethylene glycol (PEG) linker with two (6) to three units (7), with maximum degradation (Dmax) values of 61% and 58% in HCC-827 cells, respectively. From the results obtained from in vivo studies carried out with the best compounds, a tumor growth inhibition (TGI) up to 57.35% was observed74.
In 2023, Zhu et al. published a series of anti-bromodomain-containing protein 4 (BRD4) PROTACs for the treatment of hematological malignancies. The BRD4 protein belongs to the bromodomain-containing (BRD) and extra terminal domain (BET) family, which are known to function as epigenetic readers that regulate the expression of several genes, including tumor-associated genes and anti-apoptotic genes. From the results of the anti-proliferative studies performed, PROTAC 8 was the most potent in MM.1S (half-maximal inhibitory concentration (IC50) value of 0.027 µM) and MV-4-11 (IC50 = 0.003 µM) cell lines, degrading BRD4 in a time- and concentration-dependent depletion manner. The forced degradation of BRD4 also resulted in a reduction in the expression of the c-MYC protein. Furthermore, it has shown promise in the treatment of pulmonary fibrosis, as it satisfactorily inhibits the activation of MRC5 cells by downregulating the BRD4 protein. It should also be noted that replacing pomalidomide with 4-hydroxylthalidomide reduced the antiproliferative activity in vitro, demonstrating that the chosen cereblon ligand is not indifferent75.
In 2023, Qin’s group reported a library of anticancer PROTACs targeting glutathione peroxidase 4 (GPX4). Few GPX4 inhibitors are currently available. However, its inhibition activates cell ferroptosis, a non-apoptotic iron-dependent mechanism of cell death, leading to an effective anticancer effect. Taking advantage of this information, several PROTACs resulting from the binding of ML162 derivatives and ligands to cereblon/VHL E3 ligases were synthesized to evaluate the impact of forced GPX4 degradation on cancer cells. Among them, the cereblon-based PROTAC 9 (GDC-11) (Dmax = 33%) is the most cytotoxic with an IC50 value of 12 µM in HT-1080 cells and is able to induce ferroptosis. VHL-based PROTACs, although having better degradative activity, did not induce any visible cytotoxicity or increased cellular peroxidation of lipids76.
A study published by Ciofi-Baffoni’s group reports on a promising cereblon-based peptidomimetic PROTAC targeting the dimeric SARS-CoV-2 3-chymotrypsin-like protease (3CLPro) protein, a key protein in viral replication. By conjugating the GC-376 ligand to a piperazine-piperidine derivative of pomalidomide, they obtained PROTAC 10, which was able to dramatically degrade the target protein in cultured cells without affecting host cell viability. However, it should be noted that the results obtained with PROTAC are inferior to those observed with inhibitors, such as GC-376 (IC50 = 0.19 µM). This study, therefore, highlights the promising therapeutic versatility of the use of cereblon-based PROTACs, not only in the treatment of malignant or neurodegenerative diseases, but also in the treatment of infectious diseases77.
Pomalidomide-based PROTACs from 3-nitrophthalic anhydride
As mentioned previously, pomalidomide can be obtained from the condensation between the glutarimide ring and 3-nitrophathalic anhydride (12), either commercially obtained or prepared from 3-nitrophthalic acid (11), thus forming 4-nitrothalidomide (13)78. The nitro group of 4-nitrothalidomide is then reduced to an amine group, producing pomalidomide, which can then be linked to different types of linkers (Scheme 2)78.

Chemical synthesis of pomalidomide-based PROTACs from 3-nitrophtalic anhydride. Reagents and conditions: a Acetic anhydride, reflux, 2 h; b tert-butyl-(2,6-dioxopiperidin-3-yl)carbamate 3, sodium acetate, acetic acid, reflux, 6 h; b1 3-aminopiperidine-2,6-dionehydrochloride, sodium acetate, acetic acid, 130 °C, 48 h; b2 3-aminopiperidine-2,6-dionetrifluoroacetate, triethylamine, THF, 80 °C, 6 h; b3 3-aminopiperidine 2,6-dionehydrochloride, sodium acetate, acetic acid, 80 °C, 12 h; c1 Palladium on Carbon (Pd/C), H2, DMF, rt, 24 h; c2 Fe, ammonium chloride, ethanol/water mixture, rt, overnight; c3 Ammonium formate, Pd/C, methanol, rt, 2 h; d 3-aminopiperidine-2,6-dionehydrochloride, triethylamine, THF, ≤20 °C, 30 min; e Pd/C, H2, 145 psi, methanol, rt, 30 min, quant.; f Methanol, reflux, 2 h; g glutamine, DMF, 80 to 87 °C, 8 h; h Pd/C, H2, 50 psi, metanol, 3 h; i CDI, acetonitrile, reflux, 4.5 h.
There are several methods for the reduction of 4-nitrothalidomide to pomalidomide. Of the various synthetic routes reported, reduction via palladium on carbon (Pd/C)-catalyzed hydrogenation is the one that presents the best yields (around 99%)79,80. Other reducing agents may be iron-ammonium chloride or Pd/C and ammonium formate, however, with lower yields78,81.
There are another alternative routes for the synthesis of pomalidomide. One such route commences with 3-nitrophtalic anhydride (12), yielding compound 3-aminopiperidine-2,6-dione hydrochloride (14)78. Subsequent reduction of this compound results in the formation of compound 15, which undergoes cyclization to yield the desired product78. This method has been observed to produce pomalidomide with good yields (>60%)78. An alternative approach involves the initiation of the reaction with 3-nitrophthalimide (16), which, upon reacting with glutamine via amination, results in the formation of compound 1778. This compound undergoes reduction and cyclization, leading to the production of pomalidomide, albeit with a low yield due to the poor chemical reduction78.
Steinebach and colleagues published a series of cereblon homo-PROTACs, i.e., a cereblon-based PROTAC targeting cereblon itself. Using 4-nitrothalidomide as the starting building block, PROTACs were synthesized comprising two pomalidomide moieties linked by different types of linker. The homodimerized PROTAC 18 was characterized as a potent and long-lasting degrader of cereblon protein at low concentrations by western blot studies (Table 3)80.
Another example of PROTACs starting from 3-nitrophthalic anhydride are the compounds published by the Gou group. Their paper reported on PROTACs targeting anti-casein kinase 2 (CK2), the serine/threonine protein kinase associated with tumor growth that is overexpressed in cancer cells. The reaction of pomalidomide with chloroacetyl chloride and then with sodium azide is followed by click-chemistry reaction to give triazole-based PROTACs. Among these, PROTAC 19 shows significant UPS-dependent degradation of the CK2 protein. By degrading the target, PROTAC 19 reduces AKT protein phosphorylation and leads to increased p53 expression81.
N-alkyl and acyl-connected pomalidomide-based PROTACs
When the cereblon ligand to be incorporated into PROTAC is pomalidomide, the most commonly reported synthetic route for coupling to the linker is via acylation reactions with activated acyl donors. Attaching the aniline group of pomalidomide to an acyl chloride-bearing linker generated in situ in THF under reflux is undoubtedly the most common route (Scheme 3)79,82,83. Note also that there is the possibility to react carboxylic acid linkers directly with pomalidomide using coupling agents such as 1-hydroxybenzotriazole (HOBt), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU), 1-hydroxy-7-azabenzotriazole (HOAt), among others. However, it is necessary to consider the possibility of the formation of the secondary product 21 resulting from the acylation of the imide nitrogen, which reduces the yield of the desired product.

Chemical synthesis of N-alkyl and acyl-connected pomalidomide-based PROTACs. Reagents and conditions: a Thionyl chloride, DMF or THF, reflux, 4 h; b THF, reflux, various times depending on the type of linker (4 h to overnight); c1 HOBT, EDC, triethylamine, THF or DMF, rt, 24 h (time varies depending on the type of linker); c2 HATU, DIPEA, DMF, rt, 24 h (time varies depending on the type of linker).
Besides the acylation of pomalidomide, another possibility is to perform linker attachment by N-alkylating the amine group of pomalidomide using alkylhalide linkers (Scheme 4)84. Unfortunately, this strategy is not only poorly chemoselective, but also has low chemical yields due to low nucleophilicity71.

Chemical synthesis of N-alkyl connected pomalidomide-based PROTACs starting with pomalidomide. Reagents and conditions: a DIPEA, N-methyl-2-pyrrolidone (NMP), 110 °C, 6 h; b DIPEA, DMF, rt, 16 h.
In 2023, Wu et al. reported a series of cereblon-based PROTACs targeting heat shock protein 90 isoform α (HSP90α) for the treatment of breast cancer. HSP90 is a widely expressed molecular chaperone protein responsible for maintaining the correct folding of various proteins and is therefore associated with the stability and activation of oncoproteins. It is also known that the α-isoform of HSP90 is overexpressed in invasive tumors and is associated with metastasis. Given its role in tumor development and the fact that most HSP90 inhibitors are highly toxic, the therapeutic potential of degradation via PROTACs was investigated. In vitro studies have shown that PROTAC 22, which results from the attachment of pomalidomide via an alkyl linker to the AT13387 inhibitor derivative, selectively degrades HSP90α compared with other isoforms, with a degradation potency in the micromolar range (half-maximal target degradation (DC50) value of 0.295 μM, Dmax = 66.4%). In vivo studies showed a tumor inhibition rate of 53.99%, higher than the parent inhibitor AT13387 (43%), with reduced toxicity. Pharmacokinetic (PK) assays in rats showed that PROTAC 22 has a plasma half-life of 150 min and a maximum plasma drug concentration (Cmax) of 302 ng/mL after a 15 mg/kg dose (Table 4)85.
Meng’s group (2023) investigated the ability of PROTACs to degrade extracellular signal-regulated protein kinases 1/2 (ERK1/2). Since these proteins regulate the growth, proliferation and differentiation of cells, their degradation is seen as a way to combat cancer cells. Of the various PROTACs obtained, which recruit different E3 ligases, the cereblon-based PROTACs showed the best degradation activity. The best compound was the pomalidomide-based PROTAC 23, which was able to degrade targets with nanomolar potency (DC50 value of 102 nM) and inhibit cell proliferation (IC50 value of 2.2 µM) of HCT116 cancer cells. PROTAC 23 also demonstrated the ability to inhibit tumor cell migration. In vivo xenograft studies showed significant tumor regression after treatment with this degrader, exceeding the results obtained with 5-fluorouracil86.
In 2024, Fang et al. discovered a selective long-term hypoglycemic pomalidomide-based PROTAC targeting protein tyrosine phosphatase 1B (PTP1B), with enormous potential for the treatment of PTP1B-mediated insulin sensitivity. The PTP1B protein is a non-receptor protein tyrosine phosphatase that plays a negative regulatory role in several intracellular signaling pathways, including insulin and leptin signaling. By inhibiting the phosphatase action of PTP1B on the insulin receptor (IR) and insulin receptor substrates (IRS) proteins, insulin sensitivity is increased, and consequently better glycemic control is generated. Unfortunately, the discovery of inhibitors for this promising therapeutic target for diabetes has been hampered by problems of bioavailability and selectivity. The authors therefore investigated the biological activity of a library of different anti-PTP1B degraders obtained by acylating pomalidomide via the acid chloride linker, followed by linkage via an amide to different PT1B inhibitors. Western blot analysis showed significant target degradation induced by the action of PROTACs in HepG2 cells. PROTAC 24 with the longer PEG-based linker (23 atoms) showed remarkable UPS-dependent degradation potency with DC50 values of 0.20 µM (48 h) and 0.05 (72 h). Additional studies in HepG2 cells further demonstrated that PROTAC 24 is selective and induces long-term hypoglycemic activity via activation of the IRS-1/PI3K/Akt signaling pathway. In addition, oral glucose tolerance test results in mice showed a significant reduction in blood glucose (29%)87.
In 2024, Chen and co-workers developed cereblon-based PROTACs that target the degradation of cyclin-dependent kinases 4 and 6 (CDK4/6). The CDK6 protein is often involved in controlling the G1/S transition of the cell cycle and is overexpressed and hyperactivated in several types of cancer, ultimately promoting uncontrolled cell growth of cancer cells. Several ATP-competitive CDK6 inhibitors (e.g., palbociclib, ribociclib) have been reported, but these are not very selective and are associated with serious side effects, such as neutropenia. Furthermore, the emergence of resistance is one of the shortcomings associated with the use of inhibitors. Different degraders have been reported resulting from the binding of the CDK6 ligand YY173 to pomalidomide with different types of linkers, via N-alkylation or acylation of the aniline group. In vitro degradation studies have shown that PROTAC 25 is able to rapidly induce the degradation of CDK6 (DC50 = 2.5 nM, Dmax = 96%) and CDK4 (DC50 = 10.5 nM, Dmax = 100%) proteins. By promoting the degradation of the target protein, PROTAC 25 inhibits cell proliferation of jurkat cells (IC50 value of 0.18 µM), induces apoptosis and causes cell cycle arrest in the G1 phase in a dose-dependent manner. Notably, the cell inhibition results exceed those obtained with the isolated parent inhibitor (IC50 = 1.47 µM) or palbociclib (IC50 = 1.50 µM)88.
Taking advantage of the benefits presented by induced protein degradation, Hulme et al. designed and synthesized a first-in-class dual-specificity tyrosine phosphorylation-regulated kinase (DYRK1) PROTAC. DYRK1 is a protein kinase involved in signaling pathways that promote cell proliferation and differentiation, so its deregulation is associated with the promotion of tumor growth. Overactivation of this protein is also associated with the development of neurodegenerative diseases, such as Alzheimer’s disease, as well as cognitive deficit. DYRK1A has also been identified as a target of interest for the treatment of inflammatory, metabolic, cardiovascular and infectious diseases. In this paper, the authors linked the ATP-competitive inhibitor DYRK1 to pomalidomide or a VHL ligand via various PEG-based linkers. Cereblon-based PROTAC 26 (DYR684) induced rapid, potent and selective UPS-dependent degradation of the DYRK1 protein, resulting in effective suppression of the DYRK1 pathway, superior to that observed with classical type 1 DYRK1 inhibitors. In addition, the lead compound DYR684 is metabolically stable and has a pharmacokinetic profile suitable for in vivo application89.
Introduction of vectors to facilitate the construction of PROTACs
Another possible strategy is to introduce groups that act as vectors to facilitate the attachment of the cereblon ligand to the linker. For example, the reaction between 4-fluorothalidomide (2) and tert-butyl-2-aminoacetate or the reaction between pomalidomide and tert-butylbromoacetate results in the introduction of an acetamide group (33), which facilitates the attachment of a linker with an amine terminus via the formation of an amide bond (35) (Scheme 5)68,90.

Chemical synthesis of pomalidomide-based PROTACs with two-carbon spacer to facilitate linker attachment. Reagents and conditions: a DIPEA, DMF, 90 °C, 12 h; b Dichloromethane/trifluoroacetic acid (1:1), rt, 2 h; c1 i. Thionyl chloride, DMF or THF, reflux, 4 h; ii. THF, reflux, various times depending on the type of linker (4 h to overnight); c2 HOBT, EDC, triethylamine, THF or DMF, rt, 24 h (time varies depending on the type of linker); c3 HATU, DIPEA, DMF, rt, 24 h (time varies depending on the type of linker).
For example, Gong and colleagues designed and synthesized a novel anti-anaplastic lymphoma kinase (ALK) degrader, PROTAC 27, in which the piperazine moiety of the target ligand was directly linked to the acetamide group of (2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)glycine using the coupling agent HATU in DIPEA and DMF overnight. Although several small molecule inhibitors of ALK are approved with excellent therapeutic efficacy, the emergence of resistance-conferring mutations limits their clinical use. PROTACs could therefore be an alternative. In vitro studies with PROTAC 27 have shown that it is able to degrade three types of ALK mutants (NPM-ALK, EML4-ALK and F1174L mutations). In addition to its cytotoxicity and selectivity for ALK-positive cells, PROTAC 27 demonstrated potent anti-cancer activity in a xenograft mouse model, superior to that achieved with the inhibitor ceritinib91. From this study, it can be concluded that the degradation of a POI, induced by PROTAC’s forced approach to an E3 ligase, could be a promising alternative strategy to the loss of clinical efficacy of inhibitors due to the development of resistance.
Another possibility is to react pomalidomide with haloacetyl halides such as bromoacetyl chloride or chloroacetyl chloride to give compounds 36 and 37. Compounds 36 and 37 can be used for example to alkylate amine-bearing linkers. Another possible route is to form azide 38 by refluxing compounds 36 and 37 with sodium azide in acetone overnight. Compound 38 can be used as a building block for the construction of click-chemistry based PROTACs through its reaction with an alkyne terminal linker (39). Compound 38 can also be reduced to amine 40, which facilitates the attachment of the linker to the carboxylic acid terminal via the formation of an amide bond (41) (Scheme 6)81,83.

Chemical synthesis of pomalidomide derivatives for acyl-connected based PROTACs and click-chemistry PROTACs. Reagents and conditions: a bromoacetyl chloride, THF, reflux, overnight; b chloroacetyl chloride, THF, reflux, overnight; c Sodium azide, acetone, reflux, overnight; d sodium ascorbate, Copper (II) sulfate × 5 H2O, THF/H2O, rt, overnight (depending on the type of linker); e Palladium on Carbon (Pd/C), H2, methanol, rt, 6 h; f TBTU, triethylamine, DMF, 50 °C, 24 h (depending on the type of linker).
Although amide bonds are often used to link the various components of PROTACs, it should be noted that these bonds are still susceptible to hydrolytic or enzymatic cleavage, resulting in a loss of potency of the PROTAC92. In addition, this approach adds additional polar surface area that can create physicochemical properties that may hinder cell penetration92,93,94.
Thalidomide-based PROTACs
Thalidomide-based PROTACs can be obtained by incorporating 5-aminothalidomide derivatives derived from the alkylation of 5-fluorothalidomide95. Another strategy is to use 4-hydroxythalidomide as the starting building block to create ether-linked 4-hydroxythalidomide derivatives96.
5-Aminothalidomide based PROTACs
Although less commonly reported in cereblon-based PROTACs, 5-aminothalidomide derivatives are compounds that also have the ability to bind to cereblon E3 ligase. The synthesis of 5-aminothalidomide derivatives from the substrate 5-fluorothalidomide (43) is very similar to the synthetic route from the 4-fluoro analog, both in terms of reaction conditions and reagents97. Thus, from the SNAr between 5-fluorothalidomide (43) and primary amines linkers, in the presence of DIPEA and under heating, it is possible to obtain several 5-aminothalidomide derivatives (44) (Scheme 7)96,98,99. However, the yields are slightly lower than those obtained with the 4-fluoro analog due to the lower electron-withdrawing effect of the carbonyl groups in this position. It is also reported that from the reaction between 5,6-difluorothalidomide with an equimolar amount of nucleophile it is possible to obtain mono-substituted derivatives100. From the reaction between 5-fluorothalidomide (43) and propargylamine, it is possible to obtain compound 45, which can be used as a building block for the construction of click-chemistry based PROTACs.

Chemical synthesis of 5-Aminothalidomide based PROTACs. Reagents and conditions: a 3-aminopiperidine-2,6-dionehydrochloride, potassium acetate, acetic acid, 90 °C, overnight; b DIPEA, N-methyl-2-pyrrolidone (NMP), 90 °C, overnight (depending on the type of linker); c Propargylamine, DIPEA, DMF, 90 °C, 12 h; d Copper (II) sulfate × 5 H2O, sodium ascorbate, H2O/tert-butanol, rt, 16 h (depending on the type of linker).
From the condensation of 4-nitrophthalic anhydride (47) with 3-aminopiperidine-2,6-dione trifluoroacetate, followed by reduction of the product formed (48), it is possible to obtain the 5-aminothalidomide derivative (49) by an alternative route (Scheme 8)101. As previously described for pomalidomide, new derivatives can be synthesized from 5-aminothalidomide (49) by acylating the primary amine with acyl chloride linkers, or directly with a carboxylic acid terminal linker, leading to compound 50. Another option is to alkylate the amine group of 5-aminothalidomide with alkylhalide linkers102. However, similar to the synthesis of pomalidomide derivatives, there are still problems related to the lack of chemoselectivity.

Chemical synthesis of acyl-connected 5-Aminothalidomide based PROTACs. Reagents and conditions: a 3-aminopiperidine-2,6-dione trifluoroacetate, acetic acid, reflux, 2 h; b Palladium on Carbon (Pd/C), H2, rt, 20 h; c THF, reflux, 4 h (depending on the type of linker).
In 2024, three different groups published several examples of 5-aminothalidomide-based PROTACs targeting Fms-like protein tyrosine receptor kinase 3 (FLT3), an established target in acute myeloid leukemia (AML)103. The tyrosine kinase receptor FLT3 gene is frequently mutated in AML patients (20–30%), with internal tandem duplication (ITD) being the most common FLT3 mutation (25% of AML cases)104,105. Many of the mutations result in constitutive activation of FLT3, promoting proliferation, differentiation and survival of leukemic cells105. Several FLT3 protein inhibitors are approved for the treatment of AML (e.g., quizartinib, gilterinib), but loss of clinical efficacy due to the emergence of conferring resistance mutations is common, resulting in high leukemic burden and poor prognosis105.
Hu’s group reported the orally bioavailable anti-AML PROTAC 51, capable of inducing potent and selective UPS-dependent degradation of FLT3-ITD protein (DC50 of 7.4 nM in MV4-11 cells), with a long-lasting and dose-dependent effect in vitro and in vivo. In vitro studies showed that PROTAC 51 has potent cytotoxic activity against MV4-11 (IC50 = 39.9 nM) and MOLM-13 (IC50 = 169.9 nM) cells, causing cell cycle delay in the G1 phase and inducing apoptosis. With a desirable PK profile (e.g., half-life time = 3.8 h) combined with good oral bioavailability (53%), PROTAC 51 induced dramatic tumor regression in subcutaneous AML xenograft models. In vivo studies show that PROTAC 51 eliminates CD45+ CD33+ human leukemic cells and prolongs the life of the mice tested. It is also worth noting that in drug-resistant AML cells, the anti-proliferative activity of PROTAC 51 is superior to that obtained with FLT3 inhibitors (Table 5)106.
From the analysis of the SAR studies of the reported FLT3 degraders, it is possible to verify that the cereblon-based PROTAC presented better results than the VHL-based counterparts. Within the cereblon-based PROTACs, the best ones were those with azacyclic linkers (such as piperazine, piperidine, pyrrolidine, and azetidine) instead of flexible linkers106.
In that same year, Hu et al. published another anti-FLT3 PROTAC 52, which results from the binding of the inhibitor gilterinib with 5-fluorothalidomide via 4-methylpiperidine linker. Similar to PROTAC 51, PROTAC 52 exhibits robust cytotoxic activity against several FLT3-ITD mutant AML cells (IC50 = 1.66 nM) by inducing a marked UPS-dependent degradation of the FLT3-ITD protein (DC50 = 41.35 nM). PROTAC 52, by forcing the degradation of the FLT3-ITD protein, blocked the cell cycle in the G1 phase and induced apoptosis. Oral administration of PROTAC 52 in MV-4-11 xenograft models produced a good antitumor effect with tumor growth inhibition of around 77%, although lower than that obtained with the isolated use of the parent warhead gilterinib due to the poor pharmacokinetic characteristics of PROTAC (oral bioavailability of 5%). As previously observed, also in this study the use of rigid azacyclic linkers resulted in better degradation activities. Furthermore, the attachment site of the linker on thalidomide (4′ or 5′ position) is not indifferent, in which case binding to the 4′ position of thalidomide is favored. It is important to highlight that both PROTACs 51 and 52 demonstrate that PROTACs are a promising therapeutic alternative capable of overcoming the problem of drug resistance107.
Dong’s group reported also a potent thalidomide-based PROTAC 53 with a 4-methylpiperidine linker, corroborating that the use of azacyclic linkers favors the degradational activity of FLT3 degraders. PROTAC 53 induced the degradation of FLT3 protein (DC50 = 5.69 nM), including FLT3-ITD (Dmax = 77%), as well as CDK2/4/6/9 in MV4-11 cells. Of note is the degradation of CDK2 in HL-60 cells with DC50 values of 12.6 nM and Dmax of 82%. In addition, PROTAC 53 demonstrated robust cytotoxicity against a panel of AML cell lines with IC50 values ranging from 2.90 nM (MV4-11 cells) to 37.08 nM (THP-1 cells). In vivo studies with this PROTAC have not been presented108.
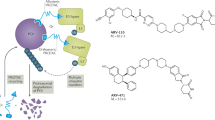
In 2024, a 5-aminothalidomide based PROTACs against the androgen receptor (AR) designated as UBX-390 (54) was published by Lee’s group. In vitro studies in prostate cancer cells with PROTAC UBX-390 (DC50 = 2.4 nM, Dmax = 96%) demonstrated that it has a prolonged degradation activity superior to that obtained with predecessor PROTACs, such as ARV-110 (DC50 = 3.1 nM, Dmax = 96%) which is currently in clinical studies. Furthermore, PROTAC UBX-390 remains active when tested in resistant prostate cancer cells, thus showing potential for the treatment of castration-resistant prostate cancer109.
4-Hydroxythalidomide based PROTACs
By exploiting the reactivity of the phenolic group of 4-hydroxythalidomide, its direct alkylation allows the synthesis of diverse and versatile starting building blocks for subsequent incorporation into the construction of cereblon-based PROTACs, e.g., via amidation, alkylation or click-chemistry reactions97,110,111,112.
Eter-connected thalidomide-based PROTACs
The synthesis of 4-hydroxythalidomide (57) can be obtained by condensation between 3-hydroxyphthalo anhydride (56) and the glutarimide ring, under different conditions and reagents (Scheme 9)110,111,112. After obtaining 4-hydroxythalidomide, it is possible to form ether bond-containing derivatives through its reaction with a linker whose terminus is iodine, bromine or tosylate group (compound 58)79,113,114,115. Since the phenolic group of 4-hydroxythalidomide can act as a nucleophile in the presence of reagents such as triphenylphosphine (TPP or PPh3) and diisopropylazodicarboxylate (DIAD) or similar, it is possible to introduce linkers bearing a primary or secondary alcohols, via Mitsunobu reactions93,116. However, it should be taken into account that although the difference in the pKa values of the two acidic groups of 4-hydroxythalidomide is sufficient to ensure selective alkylation of the phenolic group, a reaction may still occur in the imide nitrogen zone given its adequately acidity. To avoid side reactions with imide nitrogen, it is crucial to choose an appropriate base (e.g., sodium bicarbonate, sodium carbonate)117. Another possible strategy is to introduce a tert-butyloxycarbonyl (Boc)-protecting group at this position113.

Chemical synthesis of eter-connected thalidomide-based PROTACs. Reagents and conditions: a1 3-aminopiperidine-2,6-dionehydrochloride, potassium acetate, acetic acid, reflux, 24 h; a2 3-aminopiperidine-2,6-dionehydrochloride, triethylamine, toluene, reflux, 12 h; a3 i. 3-aminopiperidine-2,6-dione, triethylamine, DMF, reflux, 4 h; ii. DCC, reflux, 72 h; b Sodium bicarbonate, Potassium Iodide, DMF, 60 °C, 12 h; c Triphenylphosphine (PPh3), di-tert-butyl azodicarboxylate (DBAD), THF, rt, 2 h; d Sodium bicarbonate, DMF, 70 °C, 6 h; e DMF, 80 °C, 16 h. (Note: conditions may vary depending on the type of linker).
Several Cereblon-based PROTACs containing ether-linked 4-hydroxythalidomide derivatives have been reported in scientific literature (Table 6).
In 2023, Zhang’s group reported two innovative tumor hypoxia-activated PROTACs 59 and 60 targeting epidermal growth factor receptor 19 deletions (EGFRDEL19). Due to the abnormal vasculature of solid tumors, they present an environment with low oxygen levels (hypoxic environment), which makes it difficult to deliver drugs to the site of action and promotes metastasis of cancer cells. Thus, the present group incorporated hypoxia-activated leaving groups (HALGs), such as (1-methyl-2-nitro-1H-imidazol-5-yl)methyl or 4-nitrobenzyl groups, into ether-linked thalidomide-based PROTACs. Because HALGs were added to the nitrogen of the imide group, these PROTACs are inactive under normoxic conditions and are unable to recruit the cereblon protein. Under hypoxic conditions, the HALGs are cleaved and the PROTAC becomes active, degrading the EGFRDEL19 protein. In vitro studies have shown that PROTACs 59 and 60 satisfactorily degrade the protein of interest. Both PROTACs exhibited selective cytotoxicity dependent on hypoxia (IC50 values of 1.09 and 0.61 µM, respectively) and normoxia (IC50 values of 1.92 and 1.10 µM, respectively). Furthermore, in hypoxia these PROTACs inhibited tumor migration and induced cancer cells apoptosis. In short, this article validates an innovative and promising strategy to control the action of PROTACs, in which introducing cleavable groups under certain physiological conditions helps to increase selectivity and consequently reduce off-target effects of PROTACs118.
More recently, Ding et al. (2024) reported a novel, potent and selective thalidomide-based PROTAC for the induction of FLT3 protein degradation. In vitro studies showed that PROTAC 61 (LWY713) exhibited nanomolar degradation potency (DC50 = 0.64 nM, Dmax = 95%) in FLT3-ITD AML MV4-11 cells. The FLT3 protein degradation induced by PROTAC LWY713 results in a potent cytotoxic effect (IC50 value of 1.50 nM), as well as G0/G1 cell cycle arrest and apoptosis of MV4-11 cells. In addition, PROTAC LWY713 was shown to be a promising lead degrader with potent anti-tumor activity in vivo, significantly reducing tumor volume in the MV4-11 cell xenograft model119.
Two-carbon spacer 4-hydroxythalidomide based PROTACs
The synthesis of 4-hydroxythalidomide derivatives with a two-carbon spacer is a strategy that allows obtaining new building blocks to facilitate the construction of cereblon-based PROTACs. By alkylating the 4-hydroxyl group via tert-butyl bromoacetate or benzyl glycolate, followed by removal of the protecting group, it is possible to obtain compound 67, with a carboxylic acid terminus that can be used for linker attachment via an amide bond, and thus facilitate late-stage linker attachment (Scheme 10)120,121,122,123. However, it is necessary to consider that the introduction of a new amide bond increases the susceptibility to hydrolysis and the topological polar surface area (TPSA), as well as contributing to the increase of hydrogen bond donor (HBD) or hydrogen bond acceptor (HBA) count.

Chemical synthesis of two-carbon spacer 4-hydroxythalidomide based PROTACs. Reagents and conditions: a1 tert-butyl 2- hydroxyacetate, triphenylphosphine (PPh3), di-tert-butyl azodicarboxylate (DTBAD), THF, 0 °C to rt, overnight; a2 tert-butyl bromoacetate, potassium iodide, potassium bicarbonate, DMF, 60 °C, 12 h; a3 tert-butyl bromoacetate, potassium bicarbonate, DMF, rt, 2 h; a4) benzyl glycolate, triphenylphosphine (PPh3), diisopropyl azodicarboxylate (DIAD), THF, 0 °C to rt, 18 h; b1 Formic acid, dichloromethane, 40 °C, overnight; b2 Trifluoroacetic acid, rt, 4 h; b3 Palladium on Carbon (Pd/C), H2, methanol, rt, 3 h, quant.; c1 i. Thionyl chloride, DMF or THF, reflux, 4 h; ii. THF, reflux, various times depending on the type of linker (4 h to overnight); c2 HOBT, EDC, triethylamine, THF or DMF, rt, 24 h (time varies depending on the type of linker); c3 HATU, DIPEA, DMF, rt, 24 h (time varies depending on the type of linker).
The alkylation of 4-hydroxythalidomide with tert-butyl bromoacetate to obtain a carboxylic acid derivative in order to facilitate the binding to the amine-terminal linker, for example, was carried out by Li’s group in order to synthesize different cereblon-based PROTACs promoting the degradation of the 20S proteasome subunit β5. All reported PROTACs demonstrated potent anti-proliferative effects against several diverse types of cancer cell lines (IC50 values between 0.16 and 86.3 µM), with a pronounced effect on the human pharyngeal squamous cell carcinoma FaDu cell line (IC50 values between 0.16 µM and 2.39 µM). Among the various compounds tested, PROTACs 62 (DC50 = 0.11 µM, Dmax = 87%) and 63 (DC50 = 0.16 µM, Dmax = 85%) stand out with the most robust in vitro cytotoxic effect against FaDu and resistant KM3/BTZ cell lines. Furthermore, this PROTAC showed in vivo activity, being well-tolerated. Of note is the ability of these compounds to induce G2/M cell cycle arrest, inhibit cell migration and induce apoptosis. Taken together, these results highlight the potential that these 20S proteasome subunit β5 PROTACs present for the treatment of pharyngeal carcinoma, and for the treatment of multiple myeloma (MM) patients resistant to the bortezomib inhibitor124.
Alkyl-connected thalidomide-based PROTACs
From the analysis of cereblon-based PROTACs, it is possible to observe a wide variety of alkyl-linked thalidomide derivatives, which include alkine-containing linkers as well as their reduced analogs110,125. In this way, it is possible to obtain derivatives with a greater or lesser degree of flexibility or rigidity between the linker and the cereblon ligand. Starting from the iodo- or bromo-thalidomide derivatives (69), it is possible to connect propargyl-containing linkers via Sonogashira coupling reaction (70), with better yields using the bromo derivative (>70%) than the iodo derivative (20–70%)110,125,126. The alkyne group of the propargyl-containing thalidomide derivative can be further reduced by Pd/C catalyzed hydrogenation (compound 71), thereby increasing the degree of flexibility of the linkage (Scheme 11)110.

Chemical synthesis of alkyl-connected thalidomide-based PROTACs. Reagents and conditions: a1 (via bromo derivative) Pd(PPh3)2Cl2, Copper (I) iodide, triethylamine, DMF, 70 °C, 3 h; a2 (via bromo derivative) Pd(PPh3)2Cl2, Copper (I) iodide, triethylamine, THF, 70 °C, 12 h; a3 (via iodo derivative) Pd(PPh3)2Cl2, Copper (I) iodide, DIPEA, THF, reflux, 4 to 24 h (time varies depending on the type of linker); b Palladium on Carbon (Pd/C), H2, ethanol, rt, 12 h.
Over the past few years, several alkyl-linked thalidomide-based PROTACs have been published in the scientific literature (Table 7).
In 2021, Yang’s group reported alkyl-connected PROTAC 72, targeting programmed cell death-ligand 1 (PD-L1) protein, as a novel cancer immunotherapy strategy127. In vitro, PROTAC 72 was shown to effectively degrade the target protein in several cancer cell lines in a time- and concentration-dependent manner (IC50 values between 16 and 61 µM)127. In vivo studies with PROTAC 72 have demonstrated that by reducing PD-L1 protein levels, it leads to facilitated chemotaxis of CD8+ T cells, culminating in significant inhibition of MC-38 tumor growth. Thus, PROTAC 72 has anticancer activity via immune activation127.
In 2024, Sheng et al. reported a library of novel JAK1-targeted PROTACs for cancer treatment, using the inhibitor momelotinib (IC50 value of 11 nM) as the targeting ligand128. Among the various compounds synthesized, the alkyl-based PROTAC 73 stands out, capable of inducing in vitro a selective and potent proteasome-dependent degradation of the target protein (DC50 value of 214 nM), resulting in a favorable antiproliferative effect (IC50 value of 1.3 µM)128. Furthermore, by degrading the JAK1 protein, PROTAC 73 has been shown to reduce the phosphorylation of STAT1 and STAT3, which reinforces the impact of this degrader on the JAK/STAT signaling pathway128.
Although several alkyl-linked thalidomide-based PROTACs have been published in scientific literature, it is often observed from SAR studies that a different type of linker linkage favors the degradation activity of the PROTAC and consequently generates better biological activity. This may be due to the lower flexibility of the alkyl-connected thalidomide-based PROTAC, which may not favor the formation of a stable ternary complex essential for the transfer of ubiquitin units from the E3 ligase to the target.
Lenalidomide-based PROTACs
Lenalidomide is a known ligand of cereblon ligase and is widely used in the construction of cereblon-based PROTACs. Lenalidomide offers advantages over the incorporation of other thalidomide derivatives in the construction of PROTACs, as the absence of a phthalimide carbonyl group confers greater chemical and metabolic stability, better physicochemical properties and lower TPSA129. In general, lenalidomide-based PROTACs induce greater degradation of the target protein when compared with their pomalidomide-based counterparts. On the other hand, there are limitations in the synthesis of lenalidomide derivatives (Table 8), since the decrease in the phthalimide ring’s electrophilicity hinders SNAr reactions. Cross-coupling reactions to attach the linker to the lenalidomide derivative can lead to hydrolysis of the cyclic imides129.
N-alkyl and acyl-connected lenalidomide-based PROTACs
As described for obtaining pomalidomide derivatives, the amine group of lenalidomide can also be alkylated by alkylhalide linkers or acylated by carboxylic acid linkers (Scheme 12). By reducing the nitro group (e.g., via Pd/C-catalyzed hydrogenation or using Pd(OH)2 or iron ammonium chloride) of the starting 2-nitrolenalidomide (74), lenalidomide can be obtained130,131. From lenalidomide, it is possible to acylate the 4-amino position of lenalidomide via acid halide or directly with terminal carboxylic acid linkers to obtain acyl-connected lenalidomide PROTACs (75)132,133. Another option is to derivatize the 4-amino position with chloride, bromine or iodine terminal linkers in DIPEA and using a solvent (e.g., DMSO or NMP) at high temperatures (>90 °C) to obtain N-alkyl-linked lenalidomide PROTACs (77)134,135,136. An alternative chemical route is the derivatization of 4- and 5-bromolenalidomide to obtain new lenalidomide derivatives. For example, new 5-aminolenalidomide derivatives (80) can be obtained in good yields by Buchwald-Hartwig amination between secondary amine terminal linkers with 5-bromolenalidomide (79)98. Unlike fluorinated pomalidomide derivatives, SNAr reactions with fluorinated lenalidomide

Chemical synthesis of N-alkyl and acyl-connected lenalidomide-based PROTACs. Reagents and conditions: a1 Palladium (II) Hydroxide, H2, dioxane, 50 to 60 °C; a2 Fe, Ammonium Chloride, water, ethanol, 80 °C, 4 h; a3 Palladium on Carbon (Pd/C), H2, methanol, DMF, quant.; b THF, reflux, various times depending on the type of linker (4 h to overnight); c1) Pyridine, phosphoryl chloride, acetonitrile, rt, 3 h (depending on the type of linker); c2 HOBT, EDC, triethylamine, THF or DMF, rt, 24 h (time varies depending on the type of linker); c3 HATU, DIPEA, DMF, rt, 24 h (time varies depending on the type of linker); d DIPEA, N-methyl-2-pyrrolidone (NMP), 110 °C, 12 h (depending on the type of linker); e Pd-PEPPSI-IHeptCl, cesium carbonate, dioxane, 100 °C, 3.5 h (depending on the type of linker).
Several research groups have published different N-alkyl and acyl lenalidomide-based PROTACs (Table 9).
Yang’s group (2024) published a new anti-EGFR lenalidomide-based PROTAC 81 capable of degrading the EGFRC797S mutant protein for the treatment of lung cancer. PROTAC 81 exhibits nanomolar degradation activity (DC50 = 10.2 nM, Dmax > 95%) against EGFRC797S as well as against EGFRDel19/T790M/C797S mutations, while sparing wild-type EGFR. PROTAC strongly reduces cell proliferation of H1975-TM cells (IC50 = 10.3 nM). Oral administration of PROTAC in H1975-TM xenograft tumor model resulted in a significant antitumor effect (TGI > 50%). Since the EGFRC797S mutant protein confers resistance to the inhibitor osimertinib, PROTAC could be a promising alternative treatment for lung cancer patients harboring this mutation137.
In 2024, Wang et al. reported a lenalidomide-based PROTAC 82 that potently and selectively degrades ataxia telangiectasia and rad3-related protein (ATR), showing promise for the treatment of AML. Specifically, PROTAC 82 degraded ATR with a DC50 value of 22.9 nM and a Dmax value of 76% in MV-4-11 cells and showed the ability to inhibit cell proliferation of several AML cells (IC50 values ranging from 0.11–1.3 µM). PROTAC 82 was also shown to significantly inhibit tumor growth in vivo AML models, outperforming the positive control (VE-821 inhibitor derivative)138.
Structurally, both PROTACs 81 and 82 present lenalidomide as a cereblon recruiting moiety linked to the linker via an amide bond137,138.
In order to increase the metabolic stability of anti-Bruton’s Tyrosine Kinase (BTK) PROTACs, Wang’s group reported a series of different compounds varying the rigid linker and different cereblon ligands. Among them, the N-alkyl-lenalidomide based PROTAC 83 stands out, inducing the UPS- and concentration-dependent degradation of BTK (DC50 = 7.0 nM, Dmax = 96.5%) without inducing the degradation of cereblon neo-substrates IKZF1/3. In addition to better results in antiproliferative assays compared to the inhibitor ibrutinib, PROTAC 83 showed good metabolic stability with a half-life time of more than 145 min98.
In 2024, Liu et al. published a potential and highly selective PROTAC against the nuclear receptor binding SET domain-containing 2 (NSD2) protein, the histone methyltransferase considered a promising cancer target. Existing NSD2 inhibitors are not very selective and not very active. Thus, Liu’s group designed PROTAC 84 (LLC0424), which degrades NSD2 with nanomolar potency (DC50 = 20 nM, Dmax = 96%) in acute lymphoblastic leukemia (ALL) RPMI-8402 cells. Antiproliferative studies demonstrate cytotoxic activity of PROTAC LLC0424 against NSD2-mutant ALL cell lines. In vivo, PROTAC LLC0424 demonstrated potent degradative activity139.
Alkyl-connected lenalidomide-based PROTACs
As observed in alkyl-connected thalidomide derivatives, alkyl-lenalidomide derivatives have also been incorporated into the construction of several cereblon-based PROTACs. When establishing the connection between the linker and lenalidomide derivative, an alkyne-type connection is often observed, or an alkane-type connection as a result of the reduction of the previous connection (Scheme 13)136,140,141,142.

Chemical synthesis of alkyl-connected lenalidomide-based PROTACs. Reagents and conditions: a Pd(PPh3)2Cl2, Copper (I) Iodide, triethylamine, DMF, 80 °C, 3 h to overnight (time varies depending on the type of linker); b1 Palladium on Carbon (Pd/C), H2, methanol/DMF, rt, 12 h (depending on the type of linker); b2 Palladium on Carbon (Pd/C), H2, ethanol, rt, 2 h (depending on the type of linker); c t-BuONO, bis(pinacolato)diboron, dibenzoyl peroxide, acetonitrile, rt, 4 h; d i. Sodium periodate, THF, water, rt, 2 h; ii. 1 M HCl, rt, 18 h; e i. tert-butyl bromoacetate, Pd(PPh3)4, cesium fluoride, dimethoxyethane, dichloromethane, reflux, 18 h; ii. Trifluoroacetic acid, dichloromethano, rt, 2 h, quant.; f HATU, DIPEA, DMF, rt, overnight (depending on the type of linker).
Using lenalidomide bromide (87) as the starting building block through the Sonogashira cross-coupling reaction, it is possible to perform linker attachment with considerable yields (>40%), depending on the type of alkyne-terminal linkers chosen to incorporate (compound 86)143,144,145. If the aim is to increase the flexibility of the linker-lenalidomide interface, there is also the possibility of reducing the alkyne-linked derivatives by Pd/C-catalyzed hydrogenation (compound 87)146.
Another strategy for obtaining alkyl lenalidomide derivatives is to convert the amino group of lenalidomide to an aryl boronic ester 88, which then undergoes oxidative hydrolysis to give compound 89. Compound 89 can be reacted with tert-butyl bromoacetate via a Suzuki cross-coupling reaction and, after ester hydrolysis, yields a carboxylic acid lenalidomide derivative (90) that can be attached to amine-containing linkers, leading to compound 91147.
In 2024, Chen and colleagues reported a series of PROTACs for estrogen receptor (ER) α-positive breast cancer through induction of CDK9 protein degradation. In in vitro studies, cereblon-based PROTAC 92 (L055) was shown to induce potent antiproliferative activity (IC50 = 29 nM in MCF-7 cells), G2/M cell cycle arrest, and decreased survival of ERα-positive breast cancer cells. In vivo biological activity evaluation showed that PROTAC L055 reduced tumor growth of T47D and MCF-7 cells in nude mice. All these effects were a result of cereblon-dependent degradation of CDK9 protein promoted by lenalidomide-based PROTAC L055 (Table 10)148.
Rao et al. published the lenalidomide-based PROTAC 93 for ibrutinib-resistant malignant lymphoma in 2023. As a result of SAR studies and computational optimization, this group developed PROTAC 93 capable of inducing the degradation of the BTKC481S mutant protein (DC50 = 3.8 nM), with a DC50 approximately five times higher than that of the first-generation BTK degrader P13I. The cytotoxic effect of PROTAC 93 was also superior to its predecessor in ibrutinib-resistant HBL-1 cells (BTKC481S). Another example of a PROTAC capable of overcoming resistance to the use of inhibitors149.
In 2023, Kaneshige and co-workers also published a alkyne-connected lenalidomide-based PROTAC for the treatment of AML. PROTAC 94 (AK-2292) targets the signal transducer and activator of transcription 5 (STAT5) protein, a transcriptional factor in the JAK/STAT signaling pathway that regulates tumor cell survival, proliferation, metastasis, and immune evasion. Thus, STAT5 has great therapeutic interest for the treatment of different types of cancer. In vitro studies with a panel of AML cells demonstrated that PROTAC AK-2292 degrades both STAT5A/B isoforms with DC50 values ranging from 0.05 to 0.41 µM, and Dmax values greater than 95%. It is also important to highlight the selectivity of PROTAC AK-2292 for STAT5 over all other STAT proteins. In vivo studies with PROTAC AK-2292 resulted in rapid degradation of the target protein, and consequently in a robust dose-dependent reduction of tumor growth at well-tolerated doses. In another study published by this group, PROTAC AK-2292 demonstrated potent cytotoxic activity against a panel of chronic myeloid leukemia (CML) cells, with IC50 values around 0.2 to 2.2 μM, as well as potent antitumor activity in xenograft tumor tissues of human CML NCO2 and KU812 cell lines in mice. Note the complete depletion of STAT5 protein a few hours after administration of the degrader150.
Phenyl-substituted isoindolinone ligands for PROTAC’s construction
In 2023, Tang and colleagues reported a library of C4-phenyl lenalidomide derivatives resulting from the direct attachment of ortho-, meta- and para-substituted phenyl groups via an optimized Suzuki cross-coupling reaction (Scheme 14)129. The authors first synthesize the key iodo-lenalidomide intermediate (95) and then replace the iodide by a palladium-catalyzed coupling reaction with a mild base using a boronic ester functional group, isolating the desired products (97) using flash column chromatography129. In this way, they obtained a series of new lenalidomide derivatives, without verifying the hydrolysis of the cyclic imides. Furthermore, they reported a partial PROTAC library resulting from the ligation of small linkers with terminal benzaldehyde, which can subsequently react with hydrazide group in a 1:1 ratio thus forming acyl-hydrazone linkage PROTACs129. Since the only by-product is water, and the solvent is DMSO, the resulting PROTACs can be studied directly in biological studies. Furthermore, if the aim is to improve stability and drug-like properties, the acyl-hydrazone bond can be converted to an amide bond129.

Chemical synthesis of C4-phenyl lenalidomide derivatives. Reagents and conditions: a PdCl2(dppf) (5 mol%), potassium acetate (3 eq), DMF (0.2 M), 100 °C.
In another study, Tang et al. reported new isoindolinone compounds substituted with aromatic rings or saturated six-membered rings151. From the analysis of structure-activity relationship studies, it can be concluded that the introduction of a 4-phenyl group contributes to the binding to cereblon. In addition to being cell permeable, these new ligands did not induce degradation of the known cereblon neo-substrate IKZFs151.
Other cereblon-binding scaffolds based PROTACs
Following in vitro and in vivo studies with IMiDs and their respective IMiDs-based PROTACs, their instability has been verified, undergoing hydrolysis and racemization in the majority of culture media used and in body fluids152,153. Although the event-driven mechanism of PROTACs may mitigate the impact of compound instability, it can still completely obliterate cellular effects of PROTACs.
The four carbonyl groups of thalidomide have different pH-dependent susceptibilities to hydrolysis, with the half-life of thalidomide decreasing with increasing pH (pH > 7)152. The hydrolysis of amide linkages of phthalimido or glutarimide rings of thalidomide essentially forms products 98, 99 and 100 (Fig. 4)152,153. In addition, an intact glutarimide moiety is often observed in human plasma154.

The hydrolysis of amide linkages of phthalimido or glutarimide rings of thalidomide essentially forms compounds 98, 99, and 100.
In order to optimize physicochemical properties, increase on-target affinity and reduce instability and off-target neosubstrates, several research groups have developed novel non-phthalimide cereblon binders and incorporated them into the construction of cereblon-based PROTACs.
To synthesize new PG-based PROTACs, many research groups use the Suzuki cross-coupling reaction between 4-hydroxyphenylboronic acid and compound 101 to synthesize compound 102 (Scheme 15). The hydroxyl group of compound 102 can be alkylated with tert-butyl bromoacetate to give compound 103 which, after hydrogenation of the aromatic ring to glutarimide, gives the PG ligand 104. After removal of the protecting group, the final compound 105 is obtained. It should be noted that the yields obtained in this way are relatively high. To increase the versatility of the linkers for incorporation into cereblon-based PROTACs, it is possible to synthesize or obtain substituted phenyl derivatives (compound 106) which, after hydrogenation of the aromatic ring, give compound 107. Depending on the substituent, compound 107 can be attached to different types of linkers to construct novel cereblon-binding scaffolds based on PROTACs. Another strategy to obtain new cereblon ligands consists of coupling 3-aminopiperidine-2,6-dione hydrochloride with compound 108, via acid halide or directly through coupling agents, thus obtaining different ligands for incorporation in the construction of new cereblon-based PROTACs (compound 109).

Chemical synthesis of other cereblon-binding scaffolds based PROTACs. Reagents and conditions: a PdCl2(dppf)×CH2Cl2, K3PO4, dioxane/H2O, 100 °C, 16 h; b K2CO3, DMF, rt, 3 h; c Pd/C, H2, EtOH, RT, 16 h; d TFA, CH2Cl2, rt, 2 h; e1 10% Pd/C, triethylsilane, EtOH, 90 min; e2 H2 (15 psi), 10% Pd/C, THF, rt, 4 h; f (i) (COCl2), DMF (cat.), CH2Cl2, 0 °C, 2 h; (ii) ArCOCl, Et3N, CH2Cl2, 0 °C to rt, 18 h; g THF, 75 °C, 1 h.
In 2021, Rankovic et al. hypothesized that the electron-withdrawing phthalimide ring might favor the hydrolysis of the glutarimide ring155. Therefore, they designed a series of new and smaller cereblon ligands, phenyl glutarimide (PG) analogs. These ligands showed improved chemical stability with similar binding affinity to cereblon155. Furthermore, incorporation of these ligands into cereblon-based PROTACs has been shown to increase their potency and degradation efficacy. For example, the PG-based PROTAC 112 (SJ995973) targeting BRD4 protein (DC50 value of 0.87 nM) was shown to reduce cell viability in human AML MV4-11 cells at the order of picamolar (IC50 value of 3 pM), corroborating the advantages of incorporating PG derivatives in the construction of innovative and more stable cereblon-based PROTACs155. Unfortunately, PROTAC SJ995973 demonstrates poor in vivo PK profile (Table 11)155.
Although PG-derivatives are more stable compared to IMiDs, they are still susceptible to hydrolysis in culture media, and the C-3 of the glutarimide ring remains configurationally unstable156. In this sense, Rankovic and colleagues published new PG derivatives with not only improved chemical stability, but also without the racemization-prone chiral center in PG by replacing the glutarimide C-3 carbon with a nitrogen atom156. From the study of new phenyl dihydrouracil (PD)-based PROTACs designed by this group, PD-PROTAC 113 (SJ43489) stands out, capable of degrading the LCK protein (DC50 = 0.8 nM) and inhibiting the cell viability of KOPT-K1 cells with nanomolar potency (IC50 = 37 nM)156.
In order to optimize the physicochemical characteristics of the cereblon ligands for the development of orally bioavailable PROTACs, Rankovic’s group (2024) conducted a comprehensive in vitro/vivo comparative study with different cereblon ligands analogs157. This study resulted in the discovery of the orally bioavailable BET-PROTAC 114 (SJ44236), which in addition to having nanomolar degradative activity, presents a superior in vivo pharmacokinetic profile, with high oral bioavailability (45%) and low clearance (8.85 mL/min/kg)157.
In 2024, a paper was published by Li’s group reporting a library of novel PG-based PROTACs targeting PD-L1 protein for the treatment of breast cancer158. Among the various PROTACs studied, PROTAC 115 stands out, which was shown to be highly potent in 4T1 cells (DC50 value of 0.61 μM), inhibiting the growth of these cells, both in vitro and in vivo. From the results obtained in vivo, it is also possible to verify that the reduction in PD-L1 levels facilitates the infiltration of CD4+ T cells and CD8+ T cells into tumor, culminating in a reduction in tumor growth. This study highlights the construction of new PG-based PROTACs against a protein other than BRD4, demonstrating the versatility of application of these new cereblon ligands in the construction of new cereblon-based PROTACs.
In recent years, Hartmann’s group has reported other types of non-thalidomide cereblon ligands. In 2023, they reported a series of ortho-locked conformationally substituted benzamide derivatives that are more stable and have a reduced ability to recruit off-target neo-substrates159. Potent cereblon-based PROTACs targeting BRD4 (PROTAC 116) were obtained by incorporating benzidamide-based cereblon ligands159.
They also reported a series of 2-((hetero)aryl(methyl))thioglutarimides via the thio-Michael addition reaction and corresponding sulfone derivatives formed by oxidation of the sulfur atom, and a serie of spiro-isoxazole based analogs160,161. Overall, these ligands showed favorable lipophilicity and good binding affinities to Cereblon. Studies of PROTACs using this type of ligand have not yet been reported160,161.
Studies with tricyclic imide-based PROTACs targeting the hepatitis C virus (HCV) protease and the fibroblast growth factor receptor showed a higher affinity of these alternative ligands to cereblon compared to pomalidomide and lenalidomide58,162. Furthermore, incorporation of tricyclic imide ligands did not induce degradation of IKZF1/3 neo-substrates58,162. The synthesis of new tricyclic imide ligands can be achieved by the reaction of compound 110 with 3-aminopiperidine-2,6-dione hydrochloride, in THF under reflux (Scheme 15)58,162.
More specifically, PROTAC 117 against HCV NS3 protein efficiently degraded the target and inhibited proliferation of wild-type NS3 cells (Huh7.5 cells) with an IC50 of 748 nM. It also inhibited cell lines harboring the mutant proteins NS3V55A and NS3A156S with IC50 values of 508 nM and 1561 nM, respectively58.
Strategies such as the creation of photoswitchable PROTACs and PHOtochemically TArgeting Chimeras (PHOTACs) have been developed to control the action of PROTACs in time and space, thereby increasing their selectivity11.
Photoswitchable PROTACs have a photoswitchable module (e.g., an azobenzene group) in their linker, and the cis- and trans-configurations do not have the same activity, changing their configuration quickly and reversibly after exposure to a certain radiation163. Thus, the synthesis of cereblon ligands attached to azobenzene photoswitches linkers has been incorporated into the construction of some cereblon-based PROTACs.
There are several examples of azobenzene-based PROTACs. In the case of the anti-leukemic PROTAC 118 against Breakpoint Cluster Region Abelson (BCR-ABL) protein, there is an amide bond linking lenalidomide to the azo group. PROTAC 118 is inactive in the cis-configuration, but after irradiation with visible light it changes to the trans-configuration and is therefore active. Upon absorption of UV radiation, it reverts to its cis-configuration, and thus its inactive state. This strategy increases the precision and selectivity of the treatment and minimizes unwanted side effects133.
Another strategy to control the action of PROTACs is the incorporation of caged cereblon ligands, i.e., ligands with photocleavable groups164,165. Although different from photoswitchable PROTACs, these types of PHOTACs are not able to bind to E3 ligases when presenting this group and therefore do not induce polyubiquitination of the target protein164. However, upon exposure to a specific wavelength capable of cleaving the inactivating group, the PROTAC becomes active, resulting in the forced degradation of the target165. The nitrogen of the glutarimide ring of IMiDs has been chosen as the ideal position for the introduction of a photolabile group, such as the nitroveratryloxy-carbonyl (NVOC) or 6-nitropiperonyloxymethyl (NPOM) group164,165.
In short, all these different studies demonstrate the relevance of optimizing the ligands of E3 ligases, in this specific case of cereblon E3 ligase, for their incorporation into the construction of more potent, selective, stable PROTACs with the desirable pharmacokinetic properties for oral administration.
Cereblon-based PROTACs: non-classic synthetic approaches
As seen previously, the classical synthetic routes for obtaining PROTACs are often a time-consuming and a tedious process, with complicated purifications associated with low yields. In this sense, both academia and the pharmaceutical industry have been trying to develop innovative synthetic strategies with a view to speeding up the PROTACs’ discovery.
Click-chemistry synthesis
The term “click-chemistry” was first coined in 2001 by K. Barry Sharpless to describe a powerful set of fast, modular, efficient, stereospecific, reliable chemical reactions166.
In recent years, click-chemistry has been applied exponentially in the field of PROTAC, since the Huisgen copper-catalyzed alkyne azide cycloaddition (CuAAC) click reaction allows a variety of new triazole compounds to be obtained in a simple, rapid and high-yield manner167. Copper-catalyzed click-chemistry is performed under mild reaction conditions with stoichiometric amounts of each component (azide and alkyne building block) and with remarkable functional group compatibility168.
The debut of click-chemistry in the field of PROTACs occurred in 2017, with the publication of two papers reporting for the first time the use of CuAAC in the PROTAC synthesis169,170.
Since then, an increasing number of PROTACs based on triazole fragments have been reported in the literature. This linker typology is the result of the binding of TLM to ELM through click-chemistry reactions, i.e., a copper-catalyzed huisgen 1,3-dipolar cycloaddition reaction between azides and alkyne groups, under mild chemical conditions167. In addition to the satisfactory yields obtained, this route also offers the possibility of obtaining an enormous versatility of linkers that can be incorporated into PROTACs.
There are several ways to construct click-chemistry based PROTACs by incorporating cereblon ligands attached to various azide or alkyne terminal linkers. For example, IMiDs derivatives for click-chemistry can be constructed by SNAr reactions between amine-terminal alkine or azide-based linkers and fluorinated thalidomide derivatives, leading to compound 11983,132,171. The final coupling step of the click-chemistry building blocks, i.e., the final coupling of the alkynylated POI ligand derivative with an azide cereblon ligand is accomplished by the addition of sodium ascorbate responsible for catalyzing the reduction of copper (II) sulfate to copper (I) in situ, yielding compound 120. The click-chemistry reaction can be set up with different solvents, usually in a mixture of solvents such as water and miscible (sometimes only partially miscible) organic solvents such as tert-butyl alcohol or various alcohols (Scheme 16)125,172,173.

Chemical synthesis of click-chemistry based PROTACs. Reagents and conditions: a propargylamine, DIPEA, DMF, 90 °C, 12 h; b Copper (II) sulfate, sodium ascorbate, water/tert-butyl alcohol, rt, 16 h (depending on the type of linker).
In click-chemistry based PROTACs, the yield of the reaction depends on the linker to be incorporated (e.g., length, substitutions, binding position to the ligand)174. Although the synthesis of triazole-based PROTACs follows the principles of green chemistry, these compounds have low solubilities and are challenging from a pharmaceutical industrial viewpoint due to the difficulty of scale-up.
Several studies indicate that triazole-containing drugs are rapidly metabolized by cytochrome P450 enzymes, and although the metabolism of triazole-based PROTACs has not been extensively studied, it is expected that this may affect their bioavailability and therapeutic efficacy167,174.
Given the growing interest in the construction of click-chemistry-based PROTACs, several research groups have reported the preparation of pomalidomide, thalidomide and lenalidomide derivatives for azide-alkyne cycloaddition click reaction.
In 2017, Jung’s group reported the first-in-class triazole-based PROTAC. The cereblon-based PROTAC 121, targeting sirtuins (Sirt) histone deacetylases, was shown to selectively degrade and inhibit target activity (IC50 value of 0.25 µM), thereby inducing downstream α-tubulin hyperacetylation. Thus, this study opened the door to a new strategy for the generation of PROTACs, as well as the generation of PROTACs with potential for the treatment of neurodegenerative diseases, given the involvement of Sirt2 in Parkinson’s disease (Table 12)169.
In 2021, Sun and colleagues reported a pioneering library of cereblon-based PROTACs targeting the degradation of glycogen synthase kinase 3 (GSK-3), a protein kinase considered a therapeutic target in neurodegenerative diseases. In vitro studies demonstrated that the triazole-based PROTACs 122 promoted moderate UPS-dependent degradation of POI (Dmax > 40%), thereby inhibiting glutamate-induced cytotoxicity in HT-22 cells175.
Recently, Lie et al. also published a library of novel click-chemistry-based PROTACs recruiting cereblon, MDM2, VHL and IAP E3 ligases for PD-L1 depletion. Among the various compounds, the pomalidomide-based PROTAC 123 stands out with a potent T-cell killing capacity resulting from the forced depletion of the target protein in a dose- and time-dependent manner. This demonstrates its potential as a new strategy for tumor immunotherapy176.
One-pot synthesis
One of the approaches developed to accelerate and simplify the development of new PROTACs and minimize chemical waste by reducing the number of processing and purification steps is one-pot synthesis. The one-pot synthesis strategy consists of combining a target ligand and an E3 ligase ligand with a heterobifunctional linker, under optimized reaction conditions to assemble the desired PROTACs in good yields with minimized by-product formation. This protecting group free strategy allows the multiple reaction steps of PROTACs synthesis to be performed in a single reaction vessel, without the need to isolate intermediates71,177.
In 2021, Derksen and co-workers exploited the difference in reactivity between amine nucleophiles to synthesize for the first time pomalidomide-based PROTACs targeting BRD4 through an one-pot synthesis strategy71. Thus, they first reacted 4-flurothalidomide with an unprotected heterodiamine linker (NHR-linker-NH2), in DMSO at 50 °C to favor the reaction with the secondary amine terminal of the linker. After depletion of 4-flurothalidomide, the activated ester derivative of JQ1 was added at room temperature, which, when reacting with the primary amino terminus of the linker, directly forms the desired compound. By varying the diamine linkers, this group was able to synthesize a library of new pomalidomide-based PROTACs with good yields (20–60%), reducing steps, time, and cost71.
However, limitations of this pioneering study include the need to use heteroatom-rich amino linkers and the fact that only nucleophilic substitution reactions work well with thalidomide derivatives, making one-pot synthesis difficult to use with other types of cereblon ligands177. In order to expand the toolbox of protocols for one-pot PROTAC synthesis using building blocks with different functional groups, Krumb’s group published a new library of cereblon-based PROTACs targeting BRD4 protein via amide formation after a one-pot synthesis photoinduced C(sp2)−C(sp3) coupling (yields ranging from 7 to 93%). Various one-pot synthesis-based PROTACs with different aliphatic, aromatic and rigidified heterobifunctional linkers were synthesized, demonstrating the suitability of one-pot transformation for high-throughput screening. When tested in vitro, most of the synthesized anti-BRD4 PROTACs have shown to significantly degrade the target protein, with the most active compound (124) showing good solubility and a nanomolar degradational potency (DC50 value of 0.3 nM)177.
Multi-Component Reactions (MCRs)
In 2022, Bhela and co-workers published a paper in which they presented for the first time a multicomponent platform for the synthesis of PROTACs with different linker typologies. These PROTACs were obtained via multicomponent reactions (MCRs) entitled: four-component Ugi reaction, four-component split-Ugi type reaction with a symmetrical diamine and, a three component Passerini coupling (Fig. 5). The advantage of this approach is that through a single one-pot reaction, it is possible to quickly and easily obtain a variety of PROTACs178.

Cereblon-based PROTACs with different linker typologies can be obtained via multicomponent reactions (MCRs), such as, four-component split-Ugi type reaction with a symmetrical diamine, four-component Ugi reaction, and, a three component Passerini coupling reaction.
In the four-component Ugi reaction, the five different isocyanide derivatives of 4-hydroxythalidomide, in the presence of tritylamine or methylamine, formaldehyde, and acid-containing derivative of the target ligand (JQ1 derivative) yielded different anti-BRD4 PROTACs178. In the split-Ugi condensation, the same five isocyanide derivatives underwent reaction with the aldehyde and the acid-ligand, in the presence of piperazine as the symmetric amine178. In the Passerini reaction, only the same thalidomide-containing isocyanides with acid and formaldehyde were used. From these four one-pot MCRs, 16 anti-BRD4 PROTACs were obtained, with yields greater than 50%178. When tested in MDA-MB-231 cells, MCR-cereblon-based PROTACs were shown to satisfactorily reduce BRD4 levels178. From the analysis of SAR studies, it is possible to conclude that PROTACs from the tritylamine Ugi or piperazine split-Ugi condensation gave the highest potency (DC50 values between 60 to 240 nM) and highest Dmax (>86%). On the contrary, PROTACs with tertiary amides from the methylamine Ugi were poorly tolerated and with poor degradational activity178.
Therefore, Bhela et al. introduced a novel MCR-based platform designed to facilitate the efficient synthesis of PROTACs178. This platform’s great advantage is that it allows extensive exploration of structure-activity relationships by modifying the amine moiety in the MCR process178. This involves variations not only in linker length and composition, but also in the point of attachment to the warhead, which has impact on the affinity of the PROTACs and their stability. Importantly, this strategy offers reliability, high yields, versatility, elimination of protecting groups, stereochemical retention, and sustainability.
Solid-phase synthesis
Solid-phase synthesis (SPS) was invented in the 1960s by Robert Merrifield for the synthesis of peptides179. Nowadays, SPS has been a common strategy in synthetic chemistry labs due to its advantages, since through a single filtration of resin-bound chemicals it allows to quickly and simply separate the desired products from the by-products and intermediates existing in the reaction mixture180. The drug discovery research area has been taking advantage of the convenient application of SPS in parallel synthesis to create drug-like libraries and in high-throughput chemistry.
In 2019, Krajcovicova’s group pioneered the application of SPS in the discovery of PROTAC. In this paper, the first SPS-based PROTACs were described, which result from a single-step reaction with an on-bead cereblon ligand with kinase inhibitors of CDK4, ALK, BTK, SYK, and EGFR proteins. Thus, on the aminomethyl polystyrene-divinylbenzene (PS-DVB) resin, the link between thalidomide analogs with different linkers was established, forming the final thalidomide-preloaded resin (TPR). After obtaining thalidomide-based electrophiles, they reacted on-bead with five-amine-containing kinase inhibitors via a direct condensation reaction, in the presence of DIPEA. After a final step consisting of an acid treatment, SPS-based cereblon PROTACs are obtained with good yields (Scheme 17)181.

Chemical synthesis of solid-phase based PROTACs (Example 1). Reagents and conditions: a DBU/dichloromethane (1:1), rt, 15 min; b FAEEAA, HOBt, DIC, DMF/dichloromethane, rt, 16 h; c iodocetic acid, DIC, dichloromethane, rt, 90 min; d DIPEA, DMSO, rt, 6–16 h; e Trifluoroacetic acid/dichloromethane, rt.
In 2022, Xu et al. published a series of SPS-based PROTACs targeting hematopoietic prostaglandin D synthase (H-PGDS) and BRD4 proteins resulting from the conjugation of different chemical procedures, such as amide coupling chemistry, click-chemistry, and urea formation. In this work, an intermediate thalidomide-derivatized alkyl azide-resin 130 was first created. From this, the triazole-linked PROTAC was synthesized via azide-alkyne cycloaddition reaction, in the presence of copper (I) iodide (CuI). Amide-linked PROTACs were obtained by reducing the azide group of the resin 130 to the amine group via the Staudinger reaction, subsequently forming the amide linkage with the target ligand. Another option is the formation of urea between the amine of the intermediate and the target ligand using 4-nitrophenyl chloroformate. After the on-bead steps, an acid treatment is performed to cleavage and release the PROTACs with a high degree of purity (>95%). Biological studies with previously synthesized SPS-PROTACs demonstrated that triazole-based PROTACs were the best in degrading H-PGDS protein in KU812 cells. In the case of amide-linked PROTACs targeting the BRD4 protein, they degraded the target protein with nanomolar potency in MV4-11 cells (Scheme 18)182.

Chemical synthesis of solid-phase based PROTACs (Example 2). Reagents and conditions: a (i) Copper (I) iodide, DIPEA, DMF (ii) Trifluoroacetic acid, dichloromethane; b THPP, water/DMF; c (i) HOBt, HBTU, DIPEA, DMF (ii) Trifluoroacetic acid, dichloromethane; d (i) 4-Nitrophenyl chloromata, DMF (ii) Trifluoroacetic acid, dichloromethane.
Although practical examples of SPS-based PROTACs are still scarce and this synthesis method needs to be optimized, the results presented in these studies open doors for easier and faster development of PROTAC libraries targeting different proteins.
Direct-to-biology (D2B) approaches
The purification of synthesized compounds for biological evaluation is considered the Achilles’ heel of many medicinal chemistry programs, and even more so for PROTACs due to solubility issues. There are a number of strategies that have been published with the aim of speeding up the process of new compound discovery through the reduction of purification requirements. “Direct-to-biology” (D2B) approaches are generally defined as a set of strategies that aim at the biological study of chemical compounds without the need for post-synthesis purification, i.e., biological evaluation of the crude product183,184. Although of great interest for high-throughput screening, D2B approaches have strict requirements. Firstly, the synthesis must generate the minimum number of by-products to ensure maximum purity of the final product formed. Second, the reagents and by-products formed should not be toxic or interfere with experimental biological studies. Thirdly, the functional groups of the substrate molecule must be compatible with the reaction conditions.
The first D2B strategy applied to the development of PROTACs was reported in 2020, in which Robert et al. synthesized dozens more hydrazone-linked PROTACs targeting the ER in just a few days, via condensation between aldehydes and acyl-hydrazine ligands in DMSO184. After direct evaluation of the crude products (>90% purity by liquid chromatography–mass spectrometry (LCMS), with water as the only by-product), this group resynthesized the best compounds with bioisomeric replacement of the hydrazone by an amide linkage184.
Stability problems associated with hydrazone-based PROTACs led Tang’s group to develop the “RAPID-TAC platform” for miniaturized PROTAC synthesis using phthalimidine linkages. Several PROTACs were rapidly synthesized via dialdehyde cyclisation with amine nucleophiles (1:1 ratio in PBS buffer or DMSO at 80 °C), again with water as the sole by-product185.
The use of D2B strategies in the development of PROTACs has increased in recent years, particularly in the pharmaceutical industry. In 2022, Janssen published the multistep telescoped anti-BRD4 PROTAC synthesis for accelerated evaluation of linker effects186. In this strategy, the central point lies in the choice of mono-N-Boc-protected diamine linkers, followed by a 3-step solid-phase synthetic sequence consisting of an amide coupling, boc-group removal and another amide coupling. In the following year, AstraZeneca also reported the nanomole-scale array synthesis and direct screening of PROTAC reaction mixtures from a pre-assembled library of intermediate E3 ligase ligands attached to different linkers187. At the same time, GSK developed an integrated nanoscale D2B-PROTAC platform enabled by automation188.
So, by significantly increasing throughput, D2B platforms have gained excellent value and prominence, so research into their use in PROTACs development will undoubtedly continue to evolve.
Inactive cereblon-based PROTACs
After the discovery that thalidomide derivatives with N-alkylated glutarimide ring are unable to establish binding with cereblon E3 ligase, many research groups began to incorporate these derivatives in the construction of PROTACs with the aim of obtaining negative controls, and thus assessing whether PROTAC-induced degradation is or is not cereblon-dependent52,189.
These negative control derivatives can be obtained before or after conjugation of the glutarimide ring moiety to the phthalimide moiety (Scheme 18). In the first case, the alkylation of the imide nitrogen of the glutarimide ring is carried out only after the condensation of the glutarimide and phthalimide moieties, under conditions similar to those of the pre-conjugation alkylation (compounds 135 or 136)190. In the second case, the glutarimide moiety is first alkylated with an alkyl halide (138), such as methyl iodide, in a basic medium, and then the alkylated glutarimide is conjugated with 3-fluoro anhydride to obtain the final 4-fluorothalidomide derivative (135)80,191 (Scheme 19).

Chemical synthesis of inactive cereblon-based PROTACs. Reagents and conditions: a Methanol, Triphenylphosphine (PPh3), DIAD, THF, sonication bath, 1 h; b Ethyl iodide, potassium carbonate, acetone, reflux, 3 h; c Potassium carbonate, methyl iodide, DMF, rt, 2 h; d 3-fluorophthalic acid, sodium acetate, acetic acid, reflux, 6 h.
It is still possible to introduce alkyl groups via the Mitsunobu reaction using methanol as the methyl group donor, but the reported yields are lower80.
Cereblon-based PROTACs: factors influencing efficacy
The efficacy of cereblon-based PROTACs depends directly on the absence of mutations that interfere with the binding of the degrader to the protein, or indirectly, on mutations that interfere with the formation of the E3 ubiquitin ligase CRL4CRBN complex, such as mutations that interfere with the binding between cereblon and the DDB1 protein. Over the past few years, results from clinical trials of IMiDs in MM patients have shown that the presence of conferring resistance mutations, loss of gene copy and splice variants of cereblon are associated with resistance to IMiDs and worse clinical outcome192,193. For example, patients who are refractory to IMiDs have a higher frequency of cereblon mutations (2–10%) than untreated patients (0.5%). In addition, refractory patients have a higher rate of cereblon gene copy loss (>10%) than untreated patients (1.5%)194. The first study to demonstrate in vitro resistance to cereblon-based PROTACs showed that this was due to the downregulation of mRNA and protein, caused by a deletion of a 12 Mbp fragment of chromosome 3 containing the cereblon gene195. Another study confirmed that resistance to cereblon-based PROTACs may be caused by a mutation in exon 4 of the cereblon gene, leading to a premature stop codon in the mRNA196. Although studies with cereblon-based PROTACs are still scarce, it is expected that long-term clinical use of this type of degrader could lead to the emergence of mutations that prevent PROTAC from binding to cereblon, thus preventing poly-ubiquitination of the target protein.
As mentioned above, IMiDs are indicated for the treatment of MM. However, it is now known that cereblon expression has an impact on therapeutic response, with low expression resulting in a null or weak response to IMiDs197. Patients with high cereblon expression showed much better overall survival (OS) and progression-free survival (PFS) in response to IMiDs198,199. On the other hand, it is worth noting that the studies by Zhu and co-workers show that treatment with IMiDs leads to a decrease in cereblon expression200. Based on the results of previous studies, it is reasonable to assume that IMiDs-based PROTACs may also have their cereblon expression-dependent clinical efficacy. Therefore, depending on the expression level of cereblon protein in the target cell, the therapeutic efficacy of treatment with cereblon-based PROTACs may vary depending on the type of cancer. Long-term treatments with this type of PROTACs may lead to a loss of efficacy, due to a progressive decrease in the expression of this E3 ligase.
The efficacy of cereblon-based PROTACs also depends on the expression of other proteins that are directly or indirectly involved in the ubiquitination of the POI, such as the DDB1 and CUL4 proteins. For example, although cereblon expression is high in some cancers, a lower degradation activity of PROTACs is expected due to the lack of a DDB1/CUL4 protein. In addition, studies of resistance to cereblon-based PROTACs have shown that this may result from downregulation of transcription of E2 ubiquitin conjugating enzyme UBE2G1196. Theoretically, cereblon-based PROTACs have the potential to treat diseases in which target cells express prominent levels of not only POI but also Cereblon, DDB1 and CUL4 proteins.
The involvement of efflux proteins is known to be one of the major factors influencing the therapeutic efficacy of drugs, and this is no different in the case of PROTACs. As demonstrated by Kurimchack et al., high endogenous levels of the MDR1 protein resulted in a significant decrease in the efficacy of Cereblon-based PROTAC dBET6 when compared with PROTACs-naïve cancer cells with low expression of MDR1201. The use of multidrug resistance protein 1 (MDR1) pump inhibitors restored sensitivity of PROTAC-resistant cancer cells201. Analysis of ovarian carcinoma A1847 cells resistant to Cereblon-based PROTACs revealed an upregulation of ABCB1 gene expression201. Therefore, it is expected that the upregulation of drug efflux pump(s) of which PROTAC is a substrate will result in a reduction in the amount of degrader that reaches the target cell, and consequently in a lower polyubiquitination of the target protein.
As mentioned above, there are several factors that could cause Cereblon-based PROTACs to become less effective or fail to work. These include reduced expression of cereblon, or other proteins involved in the UPS, genetic alterations including mutations, deletions or alternative splice variants, and upregulation of drug efflux pump(s). However, there are few results on this topic. So, there is a need to explore, identify and understand the factors that may influence the effect of PROTACs, and thus find strategies to optimize their impact.
Molecular Glue Degraders (MGDs) based on Cereblon ligands
Among the various TPD modalities, molecular glue degraders (MGDs) have gained prominence over the last few years. Similar to PROTACs, MGDs are also an innovative approach that allows inducing forced degradation of a protein of interest202. Upon binding of these small molecules to a certain E3 ligase, a conformational change occurs that facilitates the binding of the E3 ligase to the POI (via induction or stabilization of protein-protein interactions), thus culminating in poly-ubiquitination and consequent proteasomal degradation of the target protein203. Historically, MGDs have been discovered serendipitously204. Most are claimed as inhibitors, but later it is discovered that they generate a neomorphic surface on E3 ligase thus inducing proximity between the E3 ligase and a neo-substrate, leading to the degradation of the latter205.
Thalidomide and its derivatives, lenalidomide and pomalidomide, are some of the most well-known MGDs identified so far203. As discussed previously, when thalidomide binds to cereblon, it redirects this E3 ligase to the IKZF1/3 proteins, causing their degradation206. From the analysis of crystal and cryo-electron microscopy (cryoEM) structural studies it was possible to confirm the role of IMiDs as MGDs, since their binding to cereblon generates a neomorphic surface that confers greater selectivity for a given neo-substrate, resulting from the establishment of IMiD-dependent interactions51,207. In addition to inducing the degradation of IKZF1/3 proteins, lenalidomide induces the degradation of the neoabstract casein kinase 1α (CK1α), resulting in its approval for the treatment of myelodysplastic syndrome (MDS)208. Pomalidomide additionally degrades the ARID2 neosubstrate209. Therefore, several researchers have studied the ability of new chemical modifications in the thalidomide skeleton to induce cereblon-dependent degradation of new neo-substrates.
In 2016, the cereblon-based MGD CC-885 was reported to be capable of inducing the degradation of the transcription factor GSPT1, and thus presenting a potent preclinical anticancer activity, more specifically an antiproliferative effect in AML cells210. Through Pd-catalyzed cyanation of 5-bromolenalidomide and consequent reduction of the nitrile group, the Celgene research group obtained intermediate 140 (Scheme 20)210. From the reaction of the benzylamine group of intermediate 140 with an isocyanate derivative, compound CC-885 was subsequently obtained, with good yields210.

Chemical synthesis of Molecular Glue Degraders (MGDs) based on Cereblon ligands. Reagents and conditions: a Pd2(dba)3, dppf, Zn(CN)2, DMF, 120 °C, 19 h; b Pd/C, H2, HCl, NMP, rt, o.n.; c Et3N, THF, 40 °C, 5 h; d K2CO3, MeCN, 50 °C, 12 h; e CH2Cl2, rt, 1 h; f KOtBu, THF, −78 °C, 30 min; g i) KOtBu, THF, −78 °C, 2 h; ii) DIPEA, MeCN, 40 °C, 18 h.
In 2018, Chamberlain’s group synthesized the new cereblon modulator CC-220 (Iberdomide), from compound 141, which when alkylated and subsequently cyclized to form the glutarimide ring, is capable of binding to cereblon with greater affinity than lenalidomide211,212. The compound CC-220 is currently in phase III clinical trials for the treatment of systemic lupus erythematosus and relapsed/refractory multiple myeloma, through the induction of IKZF1 and IKZF3 protein degradation213.
Another thalidomide derivative in clinical development is MGD CC-92480 (Mezigdomide) for the treatment of relapsed refractory multiple myeloma (phase II)214. This compound results from an extension, via the piperazine group, of the solvent-exposed side chain in CC-220215. This structural alteration results in a lenalidomide-based MGD with an increased ability to degrade the IKZF3 protein and thus generate a potent immunostimulatory effect214,215.
Compared to PROTACs, MGDs are smaller molecules, with improved drug-like properties, and which allow the degradation of unligandable target proteins, without the occurrence of the hook effect204. However, the rational design and synthesis of new MGDs is still little studied. While the number of MGDs reported in scientific literature and in clinical development (approximately 20 MGDs) remains modest, it is anticipated that this domain of TPD will expand in the coming years, driven by a deeper comprehension of the protein-protein interaction interfaces and the more rational development of new compounds203,204.
Cereblon-based PROTACs in clinical trials
According to clinicaltrials.gov database, of the approximately 25 PROTACs currently in clinical trials, nineteen recruit the cereblon E3 ligase, the vast majority of which are intended for the treatment of cancer (Table 13). Although much of the clinical trials information on cereblon-based PROTACs is not yet publicly available, it is known that most are intended for oral administration.
Arvinas, founded by Dr. Crews, is a pioneer company in the commercial development of PROTACs. Currently, it has a strong portfolio of PROTACs, with several degraders already in different phases of clinical trials. The highlights of Arvina’s pipeline are PROTAC ARV-471 (Vepdegestrant), ARV-110 (Bavdegalutamide) and ARV-766 (Luxdegalutamide). PROTAC ARV-471 is in the most advanced phase of clinical trials (Phase III) for the oral treatment of advanced or metastatic breast cancer driven by ER degradation. In 2024, ARV-471 received FDA fast-track designation for the treatment of metastatic breast cancer, further strengthening its therapeutic potential216. Phase II results showed that PROTAC ARV-471 reduced ER levels (Dmax = 89%) and demonstrated a clinical benefit rate of 38% with a good tolerability profile217,218. PROTACs ARV-110 and ARV-766 are in phase I/II and are intended for the oral treatment of prostate cancer by depleting the AR. Clinical results with PROTAC ARV-471 demonstrated potent suppression of tumor growth in patients with intrinsic and acquired resistance to enzalutamide (100% and 70%, respectively)219. Recently announced clinical data for PROTAC ARV-766 showed that this degrader is effective in patients with resistance-conferring mutations in the AR gene and has a good tolerability profile220,221.
Bristol-Myers Squibb has also invested in the development of new PROTACs, several of which are in clinical trials. Its pipeline includes the cereblon-based PROTAC CC-94676, which also targets AR and will enter phase III trials in early 2025. Clinical results published to date show good anti-tumor activity in metastatic castration-resistant prostate cancer (mCRPC) patients with a manageable safety profile222.
C4 Therapeutics, Kymera, Nurix Therapeutics, Accutar Biotech are examples of other sponsors with cereblon-based PROTACs in clinical trials, most of which are at an early stage of clinical development (phase I) and therefore clinical results from these trials are still scarce. It should be noted that some of the sponsors to date have partnerships with large pharmaceutical companies (e.g., Pfizer, Sanofi, Novartis or Merck), which highlights the significant funding attracted by this cutting-edge drug strategy, indicating that cereblon-based PROTACs have a huge promising potential.
Given the increasing number of PROTACs recruiting the cereblon E3 ligase in preclinical development, it is expected that the number of these degraders in clinical trials will increase in the coming years.
Conclusions and future perspectives
PROTACs have revolutionized the entire paradigm of drug discovery and medicinal chemistry research, attracting growing worldwide interest from both academia and the pharmaceutical industry to the extent that articles on PROTACs are published daily. It was the disruptive mechanism of action that made PROTACs, and cereblon-based PROTACs in particular, so successful. By promoting UPS-dependent, non-natural degradation of the target protein, cereblon-based PROTACs have been shown to overcome many of the challenges of occupancy-driven inhibition by SMIs. Further, cereblon-based PROTACs have opened a Pandora’s box by successfully degrading several desirable “undruggable targets” for the treatment of a wide range of diseases34.
Having reviewed the main cereblon ligands incorporated in the construction of cereblon-based PROTACs, the synthetic routes for obtaining these degraders, and having analyzed the main results of their biological evaluation, it is still important to discuss and reflect on the current challenges that cereblon-based PROTACs still face, as well as their future prospects.
There are multiple possible synthetic routes to obtain different cereblon ligand derivatives, and subsequent attachment to linkers for the construction of the final cereblon-based PROTACs. The most common chemical reactions to obtain cereblon-based PROTACs include alkylation, acylation, SNAr, Mitsunobu reactions, among others. However, classical synthetic routes are time-consuming, with complicated purifications associated with low yields due to poor chemoselectivity. In order to increase the chemical yield and minimize the formation of by-products, many groups have used strategies to protect the imide groups, such as the introduction tert-butyloxycarbonyl (Boc) or p-methoxybenzyl (PMB) as protecting groups, to avoid acylation or alkylation of the nitrogen of the imide ring, which produces inactive compounds113. Another strategy is to choose the best binding order of the different building blocks to minimize the formation of by-products under conditions that are not chemoselective for cereblon ligands. In the last few years, new strategies have been applied to accelerate PROTAC screening campaigns in order to obtain cereblon-based PROTACs faster, with high purity and good yields (e.g., click-chemistry, MCRs reactions, D2B approaches).
From the biological evaluation, both in vitro and in vivo, of the many cereblon-based PROTACs reported in recent years, it is worth highlighting that they have several advantageous characteristics, such as: (1) rapid, potent and efficient degradation of POIs, isoforms and protein aggregates; (2) they do not cause overexpression of the target protein or intracellular accumulation of POI; (3) degrade the entire target protein, eliminating enzymatic and non-enzymatic functions; (4) catalytic mechanism of action (event-driven mechanism), reducing the required therapeutic dose and off-target effects; (5) highly selective and specific degradation of POI; (6) degradation of mutant proteins that confer resistance; (7) targeting of “undruggable” protein targets; (8) desirable safety and pharmacokinetic profiles; (9) can be used alone or in combination with other therapeutic strategies. Most of the cereblon-based PROTACs are in preclinical studies, and they have shown very promising therapeutic results not only for the treatment of distinct types of cancer, but also for the treatment of cardiovascular, metabolic, neurodegenerative, immune and infectious diseases. It should also be noted that most PROTACs in clinical trials are cereblon-based PROTACs, with the majority being in phase I.
From the SAR studies, it is possible to verify that the choice of the target ligand, the linker or the cereblon ligand to be incorporated in the construction of cereblon-based PROTACs is not indifferent, nor is the way in which these components are connected to each other. This is because each of these pieces affects not only the degradation activity, but also the selectivity or stability of PROTAC. It is more important to find the best possible combination between the three different building blocks than to look at the effect of each building block individually. Although the information from SAR studies is already considerable, the development of cereblon-based PROTACs is still largely based on trial and error. However, the use of computational tools and artificial intelligence will assist in the rational design of new cereblon-based PROTACs in the near future.
Analysis of the structure of cereblon-based PROTACs shows that the majority incorporate alkyl or intra-oxygenated-based linkers. More recently, the effect of incorporating rigid linkers containing cyclic structures has been studied, as this results in PROTACs that are not only more potent, but also more stable. Nowadays, it is known that the linker is an essential part to obtain the best possible PROTAC, since it influences its geometry and consequently aspects such as solubility, permeability or metabolic stability. In regard to cereblon ligands, derivatives of pomalidomide, thalidomide, and lenalidomide are most commonly incorporated into cereblon-based PROTACs. However, due to instability issues, several research groups have attempted to incorporate more stable and smaller ligands. The future goal is to incorporate better ligands for a robust degradation performance associated with better pharmacokinetic (with good oral bioavailability) and safety profiles, since the choice of ligand affects the ability of the PROTAC to induce degradation of neo-substrates, thus contributing to the occurrence of adverse effects. Therefore, it is highly demanding to optimize all components in order to obtain the best cereblon-based PROTACs.
One of the major challenges in developing novel PROTACs is achieving compounds with favorable oral pharmacokinetic properties. PROTACs typically occupy the beyond-Rule-of-Five (bRo5) chemical space, characterized by high molecular weights (700–1200 Da), elevated lipophilicity, and suboptimal numbers of rotatable bonds (nRotBs), polar surface area, hydrogen bond donors (HBD), and acceptors (HBA)223,224. These physicochemical features can adversely affect solubility and permeability, which limits oral absorption and metabolic stability. However, several orally bioavailable PROTACs have been reported, most of them recruiting the cereblon E3 ligase, with some advancing into clinical studies (e.g., ARV-110 or ARV-471). Cereblon-based PROTACs are considered the most ‘oral drug-like’ due to their cereblon ligase ligands. Pomalidomide and lenalidomide, for example, have good oral pharmacokinetic properties, including low molecular weight, acceptable HBD/HBA counts and good lipophilicity. VHL ligands, on the other hand, have a high molecular weight and are bulky206. Furthermore, optimizing TLM and linkers can reduce the molecular weight of cereblon-based PROTACs below 700 Da, thereby favoring their oral absorption and metabolic stability. Nevertheless, further research is needed to develop new orally bioavailable cereblon-based PROTACs. Among future strategies, optimizing the molecular properties of cereblon-based PROTACs stands out as a key focus. This involves refining both the target ligands and cereblon ligands - an approach exemplified by the work of Rankovic’s or Tang’s research groups—through the development of smaller, more stable cereblon binders with improved oral pharmacokinetics129,155,157. For linker design, the use of shorter, more rigid, all-carbon linkers, as opposed to flexible or PEG-based ones, has been shown to enhance permeability, stability, and reduce efflux susceptibility225. Additionally, the integration of machine learning, in silico modeling, and artificial intelligence is expected to accelerate and guide the development of orally bioavailable cereblon-based PROTACs226,227.
To date, there are few studies aimed at evaluating the effects of long-term therapy with cereblon-based PROTACs, the factors affecting therapeutic efficacy, and the emergence of resistance to their use. Therefore, results from clinical trials are expected to provide a better understanding of the pharmacokinetics and pharmacodynamics of cereblon-based PROTACs and guide future optimization of their design and development.
The results achieved with cereblon-based PROTACs have opened a new chapter in drug discovery research. The therapeutic possibilities offered by this type of degrader are numerous and relevant. However, a deeper understanding of cereblon-based PROTACs is needed in various aspects, starting from their design and synthesis to their clinical evaluation. Therefore, future efforts are needed to achieve greater rationalization of their design, improvement of their chemical synthesis (less laborious synthetic routes, with fewer steps, good yields, minimal waste and unwanted products), and optimization of their pharmacokinetic/pharmacodynamic profile. In conclusion, although there is still a long way to go and many challenges to overcome, cereblon-based PROTACs have proven to be a very promising and powerful therapeutic weapon against a wide range of diseases.
References
Amirian, R. et al. Targeted protein modification as a paradigm shift in drug discovery. Eur. J. Med. Chem. 260, 115765 (2023).
Prabakaran, S., Lippens, G., Steen, H. & Gunawardena, J. Post‐translational modification: nature’s escape from genetic imprisonment and the basis for dynamic information encoding. WIREs Syst. Biol. Med. 4, 565–583 (2012).
Lee, J. M., Hammarén, H. M., Savitski, M. M. & Baek, S. H. Control of protein stability by post-translational modifications. Nat. Commun. 14, 201 (2023).
Kim, H. K. et al. Cereblon: promise and challenges for combating human diseases. Pflügers Arch. Eur. J. Physiol. 473, 1695–1711 (2021). This article highlights the critical role of cereblon in disease treatment, emphasizing its therapeutic potential in cancer and other disorders, while addressing the challenges in optimizing drug efficacy and safety.
Pohl, C. & Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366, 818–822 (2019).
Wang, Y. & Le, W. D. Autophagy and ubiquitin-proteasome system. In Autophagy: Biology and Diseases. Adv. Exp. Med. Biol. 1206, 417–441 (2019).
Choudhary, D. et al. Target protein degradation by protacs: a budding cancer treatment strategy. Pharmacol. Ther. 250, 108525 (2023).
Burslem, G. M. & Crews, C. M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 181, 102–114 (2020).
Kleiger, G. & Mayor, T. Perilous journey: a tour of the ubiquitin–proteasome system. Trends Cell Biol. 24, 352–359 (2014). Explores the complexities of the ubiquitin-proteasome system, crucial for regulating protein degradation. It emphasizes its significance in cellular function, disease mechanisms, and potential therapeutic targets.
Park, J., Cho, J. & Song, E. J. Ubiquitin–proteasome system (UPS) as a target for anticancer treatment. Arch. Pharm. Res. 43, 1144–1161 (2020).
Kim, Y., Kim, E.-K., Chey, Y., Song, M.-J. & Jang, H. H. Targeted protein degradation: principles and applications of the proteasome. Cells 12, 1846 (2023).
Kwon, Y. T. & Ciechanover, A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci. 42, 873–886 (2017).
Guedeney, N., Cornu, M., Schwalen, F., Kieffer, C. & Voisin-Chiret, A. S. PROTAC technology: a new drug design for chemical biology with many challenges in drug discovery. Drug Discov. Today 28, 103395 (2023).
Mishra, R., Upadhyay, A., Prajapati, V. K. & Mishra, A. Proteasome‐mediated proteostasis: novel medicinal and pharmacological strategies for diseases. Med. Res. Rev. 38, 1916–1973 (2018).
Nalepa, G., Rolfe, M. & Harper, J. W. Drug discovery in the ubiquitin–proteasome system. Nat. Rev. Drug Discov. 5, 596–613 (2006).
Damgaard, R. B. The ubiquitin system: from cell signalling to disease biology and new therapeutic opportunities. Cell Death Differ. 28, 423–426 (2021).
Chen, Y., Xue, H. & Jin, J. Applications of protein ubiquitylation and deubiquitylation in drug discovery. J. Biol. Chem. 300, 107264 (2024).
He, S., Dong, G., Cheng, J., Wu, Y. & Sheng, C. Strategies for designing proteolysis targeting chimaeras (PROTACs). Med. Res. Rev. 42, 1280–1342 (2022).
Sakamoto, K. M. et al. Protacs: chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 98, 8554–8559 (2001). The first to report on the development of PROTACs, marking a major innovation in the field by introducing chimeric molecules designed to induce targeted protein degradation through the Skp1–Cullin–F box complex.
Liu, J. et al. PROTACs: a novel strategy for cancer therapy. Semin. Cancer Biol. 67, 171–179 (2020).
Mi, D., Li, Y., Gu, H., Li, Y. & Chen, Y. Current advances of small molecule E3 ligands for proteolysis-targeting chimeras design. Eur. J. Med. Chem. 256, 115444 (2023).
Kelm, J. M. et al. PROTAC’ing oncoproteins: targeted protein degradation for cancer therapy. Mol. Cancer 22, 62 (2023).
Zou, Y., Ma, D. & Wang, Y. The PROTAC technology in drug development. Cell Biochem. Funct. 37, 21–30 (2019).
Kaur, S. D., Bedi, N., Kumar, D. & Kapoor, D. N. PROTACs: promising approach for anticancer therapy. Cancer Lett. 556, 216065 (2023).
Zeng, S. et al. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: recent progress and future challenges. Eur. J. Med. Chem. 210, 112981 (2021).
Dale, B. et al. Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 21, 638–654 (2021).
Wang, Y.-W. et al. PROTACS: a technology with a gold rush-like atmosphere. Eur. J. Med. Chem. 247, 115037 (2023).
Cheng, B. et al. Recent advances in developing targeted protein degraders. Eur. J. Med. Chem. 284, 117212 (2025).
Wang, X. et al. Annual review of PROTAC degraders as anticancer agents in 2022. Eur. J. Med. Chem. 267, 116166 (2024).
Tong, B. et al. A nimbolide-based kinase degrader preferentially degrades oncogenic BCR-ABL. ACS Chem. Biol. 15, 1788–1794 (2020).
Hussain, M. S. et al. Exploiting E3 ligases for lung cancer therapy: the promise of DCAF-PROTACs. Pathol. Res. Pract. 270, 156001 (2025).
Park, S. Y. et al. Development of KEAP1-targeting PROTAC and its antioxidant properties: in vitro and in vivo. Redox Biol. 64, 102783 (2023).
Cieślak, M. & Słowianek, M. Cereblon-recruiting PROTACs: will new drugs have to face old challenges? Pharmaceutics 15, 812 (2023).
Wang, C., Zhang, Y., Wu, Y. & Xing, D. Developments of CRBN-based PROTACs as potential therapeutic agents. Eur. J. Med. Chem. 225, 113749 (2021).
Vicente, A. T. S. & Salvador, J. A. R. PROteolysis-Targeting Chimeras (PROTACs) in leukemia: overview and future perspectives. MedComm 5, 1–41 (2024).
Higgins, J. J., Pucilowska, J., Lombardi, R. Q. & Rooney, J. P. A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology 63, 1927–1931 (2004).
Zhou, L. & Xu, G. The ubiquitination-dependent and -independent functions of cereblon in cancer and neurological diseases. J. Mol. Biol. 434, 167457 (2022).
Fuchs, O. Targeting cereblon in hematologic malignancies. Blood Rev. 57, 100994 (2023).
Cheng, J. et al. The emerging role for Cullin 4 family of E3 ligases in tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 1871, 138–159 (2019).
Nguyen, K. M. & Busino, L. Targeting the E3 ubiquitin ligases DCAF15 and cereblon for cancer therapy. Semin. Cancer Biol. 67, 53–60 (2020).
Iovine, B., Iannella, M. L. & Bevilacqua, M. A. Damage-specific DNA binding protein 1 (DDB1): a protein with a wide range of functions. Int. J. Biochem. Cell Biol. 43, 1664–1667 (2011).
Kim, H. K. et al. Cereblon in health and disease. Pflügers Arch. Eur. J. Physiol. 468, 1299–1309 (2016).
Ito, T. Protein degraders—from thalidomide to new PROTACs. J. Biochem 175, 507–519 (2024).
Kim, J. H. & Scialli, A. R. Thalidomide: the tragedy of birth defects and the effective treatment of disease. Toxicol. Sci. 122, 1–6 (2011).
Vargesson, N. Thalidomide‐induced teratogenesis: history and mechanisms. Birth Defects Res. C Embryo Today Rev. 105, 140–156 (2015).
Ito, T. et al. Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350 (2010).
Lopez-Girona, A. et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 26, 2326–2335 (2012).
Lu, G. et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science 343, 305–309 (2014).
Krönke, J. et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014).
Lee, H., Neri, P. & Bahlis, N. J. Cereblon-targeting ligase degraders in myeloma. Hematol. Oncol. Clin. North Am. 38, 305–319 (2024).
Fischer, E. S. et al. Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 (2014).
Mori, T. et al. Structural basis of thalidomide enantiomer binding to cereblon. Sci. Rep. 8, 1294 (2018).
Kazantsev, A. & Krasavin, M. Ligands for cereblon: 2017–2021 patent overview. Expert Opin. Ther. Pat. 32, 171–190 (2022). This article provides an overview of patents related to cereblon ligands from 2017 to 2021.
Kim, G. Y. et al. Chemical degradation of androgen receptor (AR) using bicalutamide analog–thalidomide PROTACs. Molecules 26, 2525 (2021).
Adhikari, B. et al. PROTAC-mediated degradation reveals a non-catalytic function of AURORA-A kinase. Nat. Chem. Biol. 16, 1179–1188 (2020).
Yokoo, H. et al. Development of a hematopoietic prostaglandin D synthase-degradation inducer. ACS Med. Chem. Lett. 12, 236–241 (2021).
Konstantinidou, M. et al. The tale of proteolysis targeting chimeras (PROTACs) for Leucine‐Rich Repeat Kinase 2 (LRRK2). ChemMedChem 16, 959–965 (2021).
de Wispelaere, M. et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat. Commun. 10, 3468 (2019).
Ishida, T. & Ciulli, A. E3 ligase ligands for PROTACs: how they were found and how to discover new ones. SLAS Discov. 26, 484–502 (2021).
Poongavanam, V. & Kihlberg, J. PROTAC cell permeability and oral bioavailability: a journey into uncharted territory. Future Med. Chem. 14, 123–126 (2022).
Bricelj, A., Steinebach, C., Kuchta, R., Gütschow, M. & Sosič, I. E3 ligase ligands in successful PROTACs: an overview of syntheses and linker attachment points. Front. Chem. 9, 707317 (2021).
Liu, Y. et al. Expanding PROTACtable genome universe of E3 ligases. Nat. Commun. 14, 6509 (2023).
Zografou-Barredo, N. A., Hallatt, A. J., Goujon-Ricci, J. & Cano, C. A beginner’s guide to current synthetic linker strategies towards VHL-recruiting PROTACs. Bioorg. Med. Chem. 88–89, 117334 (2023).
Sosič, I., Bricelj, A. & Steinebach, C. E3 ligase ligand chemistries: from building blocks to protein degraders. Chem. Soc. Rev. 51, 3487–3534 (2022).
Vicente, A. T. S. & Salvador, J. A. R. Proteolysis-Targeting Chimeras (PROTACs) targeting the BCR-ABL for the treatment of chronic myeloid leukemia—a patent review. Expert Opin. Ther. Pat. 33, 397–420 (2023).
Li, D., Yu, D., Li, Y. & Yang, R. A bibliometric analysis of PROTAC from 2001 to 2021. Eur. J. Med. Chem. 244, 114838 (2022). A bibliometric analysis of PROTAC research from 2001 to 2021, shedding light on the growth, trends, and key advancements in the field.
Zhang, C. et al. Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK). Eur. J. Med. Chem. 151, 304–314 (2018).
Powell, C. E. et al. Chemically induced degradation of Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 61, 4249–4255 (2018).
Olson, C. M. et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 14, 163–170 (2018). This article compares selective CDK9 inhibition and degradation, emphasizing that while inhibition temporarily halts CDK9 activity, degradation offers a more persistent and potentially more effective therapeutic approach by eliminating the target protein entirely.
Steinebach, C. et al. A MedChem toolbox for cereblon-directed PROTACs. Medchemcomm 10, 1037–1041 (2019).
Brownsey, D. K., Rowley, B. C., Gorobets, E., Gelfand, B. S. & Derksen, D. J. Rapid synthesis of pomalidomide-conjugates for the development of protein degrader libraries. Chem. Sci. 12, 4519–4525 (2021).
Posternak, G. et al. Functional characterization of a PROTAC directed against BRAF mutant V600E. Nat. Chem. Biol. 16, 1170–1178 (2020).
Chen, L. et al. Discovery of first-in-class potent and selective tropomyosin receptor kinase degraders. J. Med. Chem. 63, 14562–14575 (2020).
Zhang, F. et al. Design, synthesis, anti-tumor activity and mechanism of novel PROTACs as degraders of PD-L1 and inhibitors of PD-1/PD-L1 interaction. Bioorg. Med. Chem. 111, 117867 (2024).
Ding, M. et al. Design, synthesis, and biological evaluation of BRD4 degraders. Bioorg. Med. Chem. 78, 117134 (2023).
Cai, M. et al. Design and synthesis of proteolysis-targeting chimeras (PROTACs) as degraders of glutathione peroxidase 4. Bioorg. Med. Chem. 90, 117352 (2023).
Grifagni, D. et al. Development of a GC-376 based peptidomimetic PROTAC as a degrader of 3-chymotrypsin-like protease of SARS-CoV-2. ACS Med. Chem. Lett. 15, 250–257 (2024).
Huang, D. et al. New synthesis route for the preparation of pomalidomide. Synth. Commun. 46, 1343–1348 (2016).
Rana, S. et al. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg. Med. Chem. Lett. 29, 1375–1379 (2019).
Steinebach, C. et al. Homo-PROTACs for the chemical knockdown of cereblon. ACS Chem. Biol. 13, 2771–2782 (2018). Explores the use of Homo-PROTACs for the chemical knockdown of cereblon, offering a novel strategy to selectively degrade cereblon and potentially enhance therapeutic approaches for diseases associated with its dysfunction.
Chen, H., Chen, F., Liu, N., Wang, X. & Gou, S. Chemically induced degradation of CK2 by proteolysis targeting chimeras based on a ubiquitin–proteasome pathway. Bioorg. Chem. 81, 536–544 (2018).
Lai, A. C. et al. Modular PROTAC design for the degradation of oncogenic BCR‐ABL. Angew. Chem. Int. Ed. 55, 807–810 (2016).
Bian, J. et al. Discovery of Wogonin-based PROTACs against CDK9 and capable of achieving antitumor activity. Bioorg. Chem. 81, 373–381 (2018).
Liu, H. et al. Discovery and characterization of novel potent BCR-ABL degraders by conjugating allosteric inhibitor. Eur. J. Med. Chem. 244, 114810 (2022). The discovery of novel potent BCR-ABL degraders, created by conjugating an allosteric inhibitor.
Jiang, Q. et al. Discovery of X10g as a selective PROTAC degrader of Hsp90α protein for treating breast cancer. Eur. J. Med. Chem. 260, 115690 (2023).
Pan, P. et al. Design, synthesis, and antitumor activity evaluation of proteolysis-targeting chimeras as degraders of extracellular signal-regulated kinases 1/2. Int. J. Mol. Sci. 24, 16290 (2023).
Yang, Z. et al. Discovery of selective proteolysis-targeting chimera degraders targeting PTP1B as long-term hypoglycemic agents. J. Med. Chem. 67, 7569–7584 (2024). One of the first examples of PROTACs being used for diabetes treatment, specifically targeting PTP1B as a promising strategy for developing long-term hypoglycemic agents.
He, H. et al. Development of degraders of cyclin-dependent kinases 4 and 6 based on rational drug design. J. Med. Chem. 67, 11354–11364 (2024).
Wilms, G. et al. Discovery and functional characterization of a potent, selective, and metabolically stable PROTAC of the protein kinases DYRK1A and DYRK1B. J. Med. Chem. 67, 17259–17289 (2024).
Huang, H.-T. et al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol. 25, 88–99.e6 (2018).
Gong, L. et al. Discovery of a miniaturized PROTAC with potent activity and high selectivity. Bioorg. Chem. 136, 106556 (2023).
Bricelj, A. et al. Influence of linker attachment points on the stability and neosubstrate degradation of cereblon ligands. ACS Med. Chem. Lett. 12, 1733–1738 (2021).
Chessum, N. E. A. et al. Demonstrating in-cell target engagement using a pirin protein degradation probe (CCT367766). J. Med. Chem. 61, 918–933 (2018).
Steinebach, C. et al. Systematic exploration of different E3 ubiquitin ligases: an approach towards potent and selective CDK6 degraders. Chem. Sci. 11, 3474–3486 (2020).
Zhou, H. et al. Discovery of orally bioavailable ALK PROTACs based ceritinib against ALK positive cancers. Eur. J. Med. Chem. 279, 116827 (2024).
Wu, T. et al. Discovery of novel flavonoid-based CDK9 degraders for prostate cancer treatment via a PROTAC strategy. Eur. J. Med. Chem. 260, 115774 (2023).
Zeng, S. et al. Discovery of highly efficient CRBN-recruiting HPK1-PROTAC as a potential chemical tool for investigation of scaffolding roles in TCR signaling. Bioorg. Chem. 143, 107016 (2024).
Chen, S. et al. Discovery of novel BTK PROTACs with improved metabolic stability via linker rigidification strategy. Eur. J. Med. Chem. 255, 115403 (2023). A novel BTK PROTACs with enhanced metabolic stability, achieved through a linker rigidification strategy.
Chen, A. et al. Identification of highly efficacious PROTACs targeting BRD4 against acute myeloid leukemia: Design, synthesis, and biological evaluations. Chin. Chem. Lett. 34, 107923 (2023).
Zhou, L. et al. Discovery of ZLC491 as a potent, selective, and orally bioavailable CDK12/13 PROTAC degrader. J. Med. Chem. 67, 18247–18264 (2024).
Capitosti, S. M., Hansen, T. P. & Brown, M. L. Facile synthesis of an azido-labeled thalidomide analogue. Org. Lett. 5, 2865–2867 (2003).
Buhimschi, A. D. et al. Targeting the C481S ibrutinib-resistance mutation in Bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry 57, 3564–3575 (2018).
Lap, C. J., Abrahim, M. S. & Nassereddine, S. Perspectives and challenges of small molecule inhibitor therapy for FLT3-mutated acute myeloid leukemia. Ann. Hematol. 103, 2215–2229 (2024).
Bystrom, R. & Levis, M. J. An update on FLT3 in acute myeloid leukemia: pathophysiology and therapeutic landscape. Curr. Oncol. Rep. 25, 369–378 (2023).
Negotei, C. et al. A review of FLT3 kinase inhibitors in AML. J. Clin. Med. 12, 6429 (2023).
Wang, J. et al. Discovery of a novel orally bioavailable FLT3-PROTAC degrader for efficient treatment of acute myeloid leukemia and overcoming resistance of FLT3 inhibitors. J. Med. Chem. 67, 7197–7223 (2024).
Ye, L. et al. Discovery of a potent Gilteritinib-based FLT3-PROTAC degrader for the treatment of Acute myeloid leukemia. Bioorg. Chem. 149, 107477 (2024).
Wu, M. et al. Discovery of a potent CDKs/FLT3 PROTAC with enhanced differentiation and proliferation inhibition for AML. Eur. J. Med. Chem. 275, 116539 (2024).
Lee, S. et al. UBX‐390: a novel androgen receptor degrader for therapeutic intervention in prostate cancer. Adv. Sci. 11, 1–13 (2024).
Zhou, B. et al. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J. Med. Chem. 61, 462–481 (2018).
Jiang, B. et al. Development of dual and selective degraders of cyclin‐dependent kinases 4 and 6. Angew. Chem. Int. Ed. 58, 6321–6326 (2019).
Papatzimas, J. W. et al. From inhibition to degradation: targeting the antiapoptotic protein myeloid cell leukemia 1 (MCL1). J. Med. Chem. 62, 5522–5540 (2019).
Scott, D. E. et al. Systematic investigation of the permeability of androgen receptor PROTACs. ACS Med. Chem. Lett. 11, 1539–1547 (2020).
McCoull, W. et al. Development of a novel B-cell lymphoma 6 (BCL6) PROTAC to provide insight into small molecule targeting of BCL6. ACS Chem. Biol. 13, 3131–3141 (2018).
Robb, C. M. et al. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem. Commun. 53, 7577–7580 (2017).
Da, Y. et al. Design, synthesis, and biological evaluation of small molecule PROTACs for potential anticancer effects. Med. Chem. Res. 29, 334–340 (2020).
Liu, Z. et al. Design and synthesis of EZH2-based PROTACs to degrade the PRC2 complex for targeting the noncatalytic activity of EZH2. J. Med. Chem. 64, 2829–2848 (2021).
Cheng, W. et al. Design, synthesis and biological evaluation of the tumor hypoxia-activated PROTACs bearing caged CRBN E3 ligase ligands. Bioorg. Med. Chem. 82, 117237 (2023).
Liu, W. et al. Discovery of LWY713 as a potent and selective FLT3 PROTAC degrader with in vivo activity against acute myeloid leukemia. Eur. J. Med. Chem. 264, 115974 (2024).
Brand, M. et al. Homolog-selective degradation as a strategy to probe the function of CDK6 in AML. Cell Chem. Biol. 26, 300–306.e9 (2019).
De Dominici, M. et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood 135, 1560–1573 (2020).
Winter, G. E. et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science (80) 348, 1376–1381 (2015).
Lohbeck, J. & Miller, A. K. Practical synthesis of a phthalimide-based Cereblon ligand to enable PROTAC development. Bioorg. Med. Chem. Lett. 26, 5260–5262 (2016). This article details the practical synthesis of a phthalimide-based cereblon ligand, facilitating PROTAC development.
Wang, S. et al. Discovery of novel 20S proteasome subunit β5 PROTAC degraders as potential therapeutics for pharyngeal carcinoma and Bortezomib-resistant multiple myeloma. Bioorg. Chem. 153, 107801 (2024).
Su, S. et al. Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders. J. Med. Chem. 62, 7575–7582 (2019).
Stewart, S. G. et al. New thalidomide analogues derived through Sonogashira or Suzuki reactions and their TNF expression inhibition profiles. Bioorg. Med. Chem. 18, 650–662 (2010).
Wang, Y. et al. In vitro and in vivo degradation of programmed cell death ligand 1 (PD-L1) by a proteolysis targeting chimera (PROTAC). Bioorg. Chem. 111, 104833 (2021).
Zhang, X. et al. Discovery of a potent and selective JAK1-targeting PROTAC degrader with anti-tumor activities. Bioorg. Med. Chem. Lett. 109, 129838 (2024).
Almodóvar‐Rivera, C. M. et al. A modular chemistry platform for the development of a cereblon E3 ligase‐based partial PROTAC library. ChemBioChem 24, 4–8 (2023).
Chaulet, C. et al. Design, synthesis and biological evaluation of new thalidomide analogues as TNF-α and IL-6 production inhibitors. Bioorg. Med. Chem. Lett. 21, 1019–1022 (2011).
Ponomaryov, Y. et al. Scalable and green process for the synthesis of anticancer drug lenalidomide. Chem. Heterocycl. Compd. 51, 133–138 (2015).
Zhang, J. et al. Development of small-molecule BRD4 degraders based on pyrrolopyridone derivative. Bioorg. Chem. 99, 103817 (2020).
Jin, Y.-H. et al. Azo-PROTAC: novel light-controlled small-molecule tool for protein knockdown. J. Med. Chem. 63, 4644–4654 (2020). Introduces Azo-PROTAC, a novel light-controlled small-molecule tool for protein knockdown. It offers a unique approach to spatially and temporally regulate protein degradation, advancing targeted therapeutic strategies.
Fischer, P. D. et al. A biphenyl inhibitor of eIF4E targeting an internal binding site enables the design of cell-permeable PROTAC-degraders. Eur. J. Med. Chem. 219, 113435 (2021).
Qu, X. et al. Effective degradation of EGFRL858R + T790M mutant proteins by CRBN-based PROTACs through both proteosome and autophagy/lysosome degradation systems. Eur. J. Med. Chem. 218, 113328 (2021).
Ren, C. et al. Structure-based discovery of SIAIS001 as an oral bioavailability ALK degrader constructed from Alectinib. Eur. J. Med. Chem. 217, 113335 (2021).
Zhu, Y. et al. Design, synthesis, and biological evaluation of novel EGFR PROTACs targeting C797S mutation. J. Med. Chem. 67, 7283–7300 (2024).
Wang, Y. et al. Discovery of selective and potent ATR degrader for exploration its kinase‐independent functions in acute myeloid leukemia cells. Angew. Chemie Int. Ed. 63, e202318568 (2024).
Liu, L. et al. Discovery of LLC0424 as a potent and selective in vivo NSD2 PROTAC degrader. J. Med. Chem. 67, 6938–6951 (2024).
Qin, C. et al. Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the bromodomain and extra-terminal (BET) proteins capable of inducing complete and durable tumor regression. J. Med. Chem. 61, 6685–6704 (2018).
Bai, L. et al. Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 77, 2476–2487 (2017).
You, I. et al. Discovery of an AKT degrader with prolonged inhibition of downstream signaling. Cell Chem. Biol. 27, 66–73.e7 (2020).
Wang, B. et al. Development of selective small molecule MDM2 degraders based on nutlin. Eur. J. Med. Chem. 176, 476–491 (2019).
Li, Y. et al. Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J. Med. Chem. 62, 448–466 (2019).
Wang, B. et al. Development of MDM2 degraders based on ligands derived from Ugi reactions: lessons and discoveries. Eur. J. Med. Chem. 219, 113425 (2021).
Li, M.-X. et al. Degradation versus inhibition: development of proteolysis-targeting chimeras for overcoming statin-induced compensatory upregulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J. Med. Chem. 63, 4908–4928 (2020).
Xiao, D. et al. Design and synthesis of new lenalidomide analogs via Suzuki cross‐coupling reaction. Arch. Pharm. (Weinheim). 353, (2020). This article explores the design and synthesis of novel lenalidomide derivatives through the Suzuki cross-coupling reaction.
Chen, W. et al. CDK9 targeting PROTAC L055 inhibits ERα-positive breast cancer. Biomed. Pharmacother. 177, 116972 (2024).
Sun, Y. et al. Developing potent BTKC481S PROTACs for ibrutinib-resistant malignant lymphoma. Chin. Chem. Lett. 34, 107924 (2023).
Kaneshige, A. et al. Discovery of a potent and selective STAT5 PROTAC degrader with strong antitumor activity in vivo in acute myeloid leukemia. J. Med. Chem. 66, 2717–2743 (2023).
Nie, X. et al. Development of phenyl-substituted isoindolinone- and benzimidazole-type cereblon ligands for targeted protein degradation. ChemBioChem 25, (2024).
Schumacher, H., Smith, R. L. & Williams, R. T. The metabolism of thalidomide: the spontaneous hydrolysis of thalidomide in solution. Br. J. Pharmacol. Chemother. 25, 324–337 (1965).
Schumacher, H., Smith, R. L. & Williams, R. T. The metabolism of thalidomide: the fate of thalidomide and some of its hydrolysis products in various species. Br. J. Pharmacol. Chemother. 25, 338–351 (1965).
Li, M., Zhi, Y., Liu, B. & Yao, Q. Advancing strategies for proteolysis-targeting chimera design. J. Med. Chem. 66, 2308–2329 (2023).
Min, J. et al. Phenyl‐glutarimides: alternative cereblon binders for the design of PROTACs. Angew. Chem. Int. Ed. 60, 26663–26670 (2021). Phenyl-glutarimides as alternative cereblon binders for PROTAC design, emphasizing their increased stability compared to traditional ligands, which could improve the efficacy and duration of targeted protein degradation therapies.
Jarusiewicz, J. A. et al. Phenyl dihydrouracil: an alternative cereblon binder for PROTAC design. ACS Med. Chem. Lett. 14, 141–145 (2023).
Actis, M. et al. Evaluation of cereblon-directing warheads for the development of orally bioavailable PROTACs. J. Med. Chem. 995973, 3591–3611 (2025).
Zhang, H. et al. Discovery of novel proteolysis-targeting chimera molecules as degraders of programmed cell death-ligand 1 for breast cancer therapy. J. Med. Chem. 67, 10589–10600 (2024).
Steinebach, C. et al. Leveraging ligand affinity and properties: discovery of novel benzamide-type cereblon binders for the design of PROTACs. J. Med. Chem. 66, 14513–14543 (2023).
Krasavin, M. et al. Synthesis of novel glutarimide ligands for the E3 ligase substrate receptor Cereblon (CRBN): Investigation of their binding mode and antiproliferative effects against myeloma cell lines. Eur. J. Med. Chem. 246, 114990 (2023).
Shevalev, R. et al. Discovery and characterization of potent spiro-isoxazole-based cereblon ligands with a novel binding mode. Eur. J. Med. Chem. 270, 116328 (2024).
Du, G. et al. Discovery of a potent degrader for fibroblast growth factor receptor 1/2. Angew. Chem. Int. Ed. 60, 15905–15911 (2021).
Verma, S. & Manna, D. Controlling PROTACs with light. ChemMedChem 15, 1258–1261 (2020).
Xue, G., Wang, K., Zhou, D., Zhong, H. & Pan, Z. Light-induced protein degradation with photocaged PROTACs. J. Am. Chem. Soc. 141, 18370–18374 (2019).
Naro, Y., Darrah, K. & Deiters, A. Optical control of small molecule-induced protein degradation. J. Am. Chem. Soc. 142, 2193–2197 (2020).
Kolb, H. C., Finn, M. G. & Sharpless, K. B. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 40, 2004–2021 (2001).
Yang, C., Tripathi, R. & Wang, B. Click chemistry in the development of PROTACs. RSC Chem. Biol. 5, 189–197 (2024). This article discusses the role of click chemistry in the development of PROTACs, highlighting its efficiency in facilitating the synthesis of chimeric molecules.
Meldal, M. & Tornøe, C. W. Cu-catalyzed azide−alkyne cycloaddition. Chem. Rev. 108, 2952–3015 (2008).
Schiedel, M. et al. Chemically induced degradation of sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). J. Med. Chem. 61, 482–491 (2018).
Wurz, R. P. et al. A “click chemistry platform” for the rapid synthesis of bispecific molecules for inducing protein degradation. J. Med. Chem. 61, 453–461 (2018).
Liu, H. et al. Construction of an IMiD-based azide library as a kit for PROTAC research. Org. Biomol. Chem. 19, 166–170 (2021).
Bemis, T. A., La Clair, J. J. & Burkart, M. D. Traceless Staudinger ligation enabled parallel synthesis of proteolysis targeting chimera linker variants. Chem. Commun. 57, 1026–1029 (2021).
Yang, Y. et al. Global PROTAC toolbox for degrading BCR–ABL overcomes drug-resistant mutants and adverse effects. J. Med. Chem. 63, 8567–8583 (2020).
Pasieka, A., Diamanti, E., Uliassi, E. & Laura Bolognesi, M. Click chemistry and targeted degradation: a winning combination for medicinal chemists? ChemMedChem 18, (2023).
Jiang, X. et al. PROTACs suppression of GSK-3β, a crucial kinase in neurodegenerative diseases. Eur. J. Med. Chem. 210, 112949 (2021).
Liu, Y. et al. Design, synthesis, and evaluation of PD-L1 degraders to enhance T cell killing activity against melanoma. Chin. Chem. Lett. 34, 107762 (2023).
Arndt, C. M. et al. One-pot synthesis of cereblon proteolysis targeting chimeras via photoinduced C(sp2)-C(sp3) cross coupling and amide formation for proteolysis targeting chimera library synthesis. J. Med. Chem. 66, 16939–16952 (2023). The use of one-pot synthesis approach for cereblon-targeting PROTACs, utilizing photoinduced cross-coupling and amide formation. This method streamlines the synthesis process, allowing for the efficient generation of diverse degraders.
Bhela, I. P. et al. A versatile and sustainable multicomponent platform for the synthesis of protein degraders: proof-of-concept application to BRD4-degrading PROTACs. J. Med. Chem. 65, 15282–15299 (2022). A versatile, sustainable multicomponent platform for synthesizing protein degraders, streamlining the process and showcasing its application in BRD4-degrading PROTACs.
Merrifield, R. B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 85, 2149–2154 (1963).
Abdildinova, A. & Gong, Y.-D. Current parallel solid-phase synthesis of drug-like oxadiazole and thiadiazole derivatives for combinatorial chemistry. ACS Comb. Sci. 20, 309–329 (2018). The paper highlights the advantages of solid-phase methods in enabling efficient, scalable, and rapid production of PROTACs, streamlining the synthesis process for drug discovery.
Krajcovicova, S., Jorda, R., Hendrychova, D., Krystof, V. & Soural, M. Solid-phase synthesis for thalidomide-based proteolysis-targeting chimeras (PROTAC). Chem. Commun. 55, 929–932 (2019).
Xu, H. et al. Development of rapid and facile solid‐phase synthesis of PROTACs via a variety of binding styles. ChemistryOpen 11, (2022).
Stevens, R. et al. Innovative, combinatorial and high-throughput approaches to degrader synthesis. Chem. Soc. Rev. 53, 4838–4861 (2024). Innovative, combinatorial, and high-throughput approaches to degrader synthesis, focusing on strategies that enhance the efficiency and scalability of producing diverse PROTACs.
Thomas, R. P. et al. A direct-to-biology high-throughput chemistry approach to reactive fragment screening. Chem. Sci. 12, 12098–12106 (2021).
Guo, L. et al. A platform for the rapid synthesis of proteolysis targeting chimeras (Rapid-TAC) under miniaturized conditions. Eur. J. Med. Chem. 236, 114317 (2022).
Hendrick, C. E. et al. Direct-to-biology accelerates PROTAC synthesis and the evaluation of linker effects on permeability and degradation. ACS Med. Chem. Lett. 13, 1182–1190 (2022).
Plesniak, M. P. et al. Rapid PROTAC discovery platform: nanomole-scale array synthesis and direct screening of reaction mixtures. ACS Med. Chem. Lett. 14, 1882–1890 (2023).
Stevens, R. et al. Integrated direct-to-biology platform for the nanoscale synthesis and biological evaluation of PROTACs. J. Med. Chem. 66, 15437–15452 (2023).
Akuffo, A. A. et al. Ligand-mediated protein degradation reveals functional conservation among sequence variants of the CUL4-type E3 ligase substrate receptor cereblon. J. Biol. Chem. 293, 6187–6200 (2018).
An, Z., Lv, W., Su, S., Wu, W. & Rao, Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 10, 606–609 (2019).
Burslem, G. M. et al. Efficient synthesis of immunomodulatory drug analogues enables exploration of structure–degradation relationships. ChemMedChem 13, 1508–1512 (2018).
Gandhi, A. K. et al. Measuring cereblon as a biomarker of response or resistance to lenalidomide and pomalidomide requires use of standardized reagents and understanding of gene complexity. Br. J. Haematol. 164, 233–244 (2014).
Kortüm, K. M. et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 128, 1226–1233 (2016).
Gooding, S. et al. Multiple cereblon genetic changes are associated with acquired resistance to lenalidomide or pomalidomide in multiple myeloma. Blood 137, 232–237 (2021).
Zhang, L., Riley-Gillis, B., Vijay, P. & Shen, Y. Acquired resistance to BET-PROTACs (proteolysis-targeting chimeras) caused by genomic alterations in core components of E3 ligase complexes. Mol. Cancer Ther. 18, 1302–1311 (2019).
Ottis, P. et al. Cellular resistance mechanisms to targeted protein degradation converge toward impairment of the engaged ubiquitin transfer pathway. ACS Chem. Biol. acschembio. 9b00525https://doi.org/10.1021/acschembio.9b00525 (2019).
Schuster, S. R. et al. The clinical significance of cereblon expression in multiple myeloma. Leuk. Res. 38, 23–28 (2014).
Broyl, A. et al. High cereblon expression is associated with better survival in patients with newly diagnosed multiple myeloma treated with thalidomide maintenance. Blood 121, 624–627 (2013).
Heintel, D. et al. High expression of cereblon (CRBN) is associated with improved clinical response in patients with multiple myeloma treated with lenalidomide and dexamethasone. Br. J. Haematol. 161, 695–700 (2013).
Zhu, Y. X. et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 118, 4771–4779 (2011).
Kurimchak, A. M. et al. The drug efflux pump MDR1 promotes intrinsic and acquired resistance to PROTACs in cancer cells. Sci. Signal. 15, (2022).
Sasso, J. M. et al. Molecular glues: the adhesive connecting targeted protein degradation to the clinic. Biochemistry 62, 601–623 (2023).
Robinson, S. A., Co, J. A. & Banik, S. M. Molecular glues and induced proximity: an evolution of tools and discovery. Cell Chem. Biol. 31, 1089–1100 (2024).
Dong, G., Ding, Y., He, S. & Sheng, C. Molecular glues for targeted protein degradation: from serendipity to rational discovery. J. Med. Chem. 64, 10606–10620 (2021).
Liu, Y., Bai, J., Li, D. & Cang, Y. Routes to molecular glue degrader discovery. Trends Biochem. Sci. 50, 134–142 (2025).
Chamberlain, P. P. & Cathers, B. E. Cereblon modulators: low molecular weight inducers of protein degradation. Drug Discov. Today Technol. 31, 29–34 (2019).
Chamberlain, P. P. et al. Structure of the human Cereblon–DDB1–lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol. 21, 803–809 (2014).
Krönke, J. et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 523, 183–188 (2015).
Yamamoto, J. et al. ARID2 is a pomalidomide-dependent CRL4CRBN substrate in multiple myeloma cells. Nat. Chem. Biol. 16, 1208–1217 (2020).
Matyskiela, M. E. et al. A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature 535, 252–257 (2016).
Bjorklund, C. C. et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia 34, 1197–1201 (2020).
Matyskiela, M. E. et al. A cereblon modulator (CC-220) with improved degradation of ikaros and aiolos. J. Med. Chem. 61, 535–542 (2018).
Werth, V. P. et al. Effect of iberdomide on cutaneous manifestations in systemic lupus erythematosus: a randomized phase 2 clinical trial. J. Am. Acad. Dermatol. 92, 435–443 (2025).
Richardson, P. G. et al. Mezigdomide plus dexamethasone in relapsed and refractory multiple myeloma. N. Engl. J. Med. 389, 1009–1022 (2023).
Hansen, J. D. et al. Discovery of CRBN E3 ligase modulator CC-92480 for the treatment of relapsed and refractory multiple myeloma. J. Med. Chem. 63, 6648–6676 (2020).
Arvinas. Vepdegestrant: a PROTAC Estrogen Receptor Degrader for Breast Cancer. https://www.arvinas.com/research-and-development/estrogen-receptor/.
Pfizer. Arvinas and Pfizer Announce PROTAC® Protein Degrader ARV-471 Continues to Demonstrate Encouraging Clinical Benefit Rate in Patients with Locally Advanced or Metastatic ER + /HER2- Breast Cancer | Pfizer. https://www.pfizer.com/news/press-release/press-release-detail/arvinas-and-pfizer-announce-protacr-protein-degrader-arv?utm_source=chatgpt.com (2021).
Arvinas. Arvinas Announces ARV-471 Achieves a Clinical Benefit Rate of 38% in Evaluable Patients and Continues to Show a Favorable Tolerability Profile in its Phase 2 Expansion Trial (VERITAC) | Arvinas. https://ir.arvinas.com/news-releases/news-release-details/arvinas-announces-arv-471-achieves-clinical-benefit-rate-38?utm_source=chatgpt.com (2022).
Mullard, A. Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discov. 20, 247–250 (2021).
Petrylak, D. P. et al. A phase 2 expansion study of ARV-766, a PROTACandrogen receptor (AR) degrader, in metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 41, TPS290 (2023).
Petrylak, D. P. et al. ARV-766, a proteolysis targeting chimera (PROTAC) androgen receptor (AR) degrader, in metastatic castration-resistant prostate cancer (mCRPC): Initial results of a phase 1/2 study. J. Clin. Oncol. 42, 5011 (2024).
Rathkopf, D. E. et al. Safety and clinical activity of BMS-986365 (CC-94676), a dual androgen receptor ligand-directed degrader and antagonist, in heavily pretreated patients with metastatic castration-resistant prostate cancer. Ann. Oncol. 36, 76–88 (2025).
Zeng, S. et al. Current advances and development strategies of orally bioavailable PROTACs. Eur. J. Med. Chem. 261, 115793 (2023).
Volak, L. P. et al. Industry perspective on the pharmacokinetic and absorption, distribution, metabolism, and excretion characterization of heterobifunctional protein degraders. Drug Metab. Dispos. 51, 792–803 (2023).
Li, Y. et al. Application and challenges of nitrogen heterocycles in PROTAC linker. Eur. J. Med. Chem. 273, 116520 (2024).
Pike, A., Williamson, B., Harlfinger, S., Martin, S. & McGinnity, D. F. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug Discov. Today 25, 1793–1800 (2020).
Apprato, G. et al. Exploring the chemical space of orally bioavailable PROTACs. Drug Discov. Today 29, 103917 (2024).
Mi, D. et al. Discovery of novel BCL6-Targeting PROTACs with effective antitumor activities against DLBCL in vitro and in vivo. Eur. J. Med. Chem. 277, 116789 (2024).
Hu, J. et al. Discovery of highly potent and efficient CBP/p300 degraders with strong in vivo antitumor activity. J. Med. Chem. 67, 6952–6986 (2024).
Pavan, A. R. et al. Cereblon-recruiting proteolysis targeting chimeras (PROTACs) can determine the selective degradation of HDAC1 over HDAC3. Chem. Commun. 60, 13879–13882 (2024).
Zeng, M. et al. Exploring targeted degradation strategy for oncogenic KRASG12C. Cell Chem. Biol. 27, e6 (2020).
Xu, Y. et al. Discovery of highly potent dual GSPT1/BRD4 degraders with anti-AML activity. Eur. J. Med. Chem. 288, 117381 (2025).
ClinicalTrials.gov. A Study to Learn About a New Medicine Called Vepdegestrant (ARV-471, PF-07850327) in People Who Have Advanced Metastatic Breast Cancer. https://clinicaltrials.gov/study/NCT05654623?term=NCT05654623&rank=1.
ClinicalTrials.gov. A Study to Compare the Efficacy and Safety of BMS-986365 Versus the Investigator’s Choice of Therapy in Participants With Metastatic Castration-resistant Prostate Cancer. https://clinicaltrials.gov/study/NCT06764485?term=NCT06764485&rank=1.
ClinicalTrials.gov. Study to Evaluate the Safety and Tolerability of CC-94676 in Participants With Metastatic Castration-Resistant Prostate Cancer. https://clinicaltrials.gov/study/NCT04428788?term=NCT04428788&rank=1.
ClinicalTrials.gov. Phase 2 Study of SAR444656 in Adult Participants With Moderate to Severe Atopic Dermatitis. https://clinicaltrials.gov/study/NCT06058156?term=NCT06058156&rank=1.
ClinicalTrials.gov. A Phase 2 Study to Evaluate the Efficacy and Safety of SAR444656 Compared With Placebo in Adult Participants With Moderate to Severe Hidradenitis Suppurativa. https://clinicaltrials.gov/study/NCT06028230?term=NCT06028230&rank=1.
ClinicalTrials.gov. Trial of ARV-110 in Patients With Metastatic Castration Resistant Prostate Cancer. https://clinicaltrials.gov/study/NCT03888612?term=NCT03888612&rank=1.
ClinicalTrials.gov. Study details | A study of ARV-766 given by mouth in men with metastatic Prostate Cancer | ClinicalTrials.gov. https://clinicaltrials.gov/study/NCT05067140?term=NCT05067140&rank=1.
ClinicalTrials.gov. A study to characterize the safety, tolerability, and preliminary efficacy of CFT1946 as monotherapy and combination therapy in subjects with BRAF V600 mutant solid tumors. https://clinicaltrials.gov/study/NCT05668585?term=NCT05668585&rank=1.
ClinicalTrials.gov. A study to assess the safety and tolerability of CFT8634 in locally advanced or metastatic SMARCB1-perturbed cancers, including synovial sarcoma and SMARCB1-Null tumors. https://clinicaltrials.gov/study/NCT05355753?term=NCT05355753&rank=1.
ClinicalTrials.gov. Study details | A dose-escalation and expansion study of BGB-16673 in participants with B-cell malignancies | ClinicalTrials.gov. https://clinicaltrials.gov/study/NCT05006716?term=NCT05006716&rank=1.
ClinicalTrials.gov. Treatment of Chinese participants with B-cell malignancies with BGB-16673, a Bruton tyrosine kinase-targeted protein-degrader. https://clinicaltrials.gov/study/NCT05294731?term=NCT05294731&rank=1.
ClinicalTrials.gov. A study to investigate safety and effectiveness of BGB-16673 in combination with other agents in participants with relapsed or refractory B-cell malignancies. https://clinicaltrials.gov/study/NCT06634589?term=NCT06634589&rank=1.
ClinicalTrials.gov. A study of NX-2127 in adults with relapsed/refractory B-cell malignancies. https://clinicaltrials.gov/study/NCT04830137?term=NCT04830137&rank=1.
ClinicalTrials.gov. FHD-609 in subjects with advanced synovial sarcoma or advanced SMARCB1-loss tumors. https://clinicaltrials.gov/study/NCT04965753?term=NCT04965753&rank=1.
ClinicalTrials.gov. Safety, PK/PD, and clinical activity of KT-413 in adult patients with relapsed or refractory B-cell NHL. https://clinicaltrials.gov/study/NCT05233033?term=NCT05233033&rank=1.
ClinicalTrials.gov. A study of NX-5948 in adults with relapsed/refractory B-cell malignancies. https://clinicaltrials.gov/study/NCT05131022?term=NCT05131022&rank=1.
ClinicalTrials.gov. A study to assess BMS-986458 alone and in combination with anti-lymphoma agents in relapsed/refractory non-Hodgkin lymphomas. https://clinicaltrials.gov/study/NCT06090539?term=NCT06090539&rank=1.
ClinicalTrials.gov. A study of AC682 for the treatment of locally advanced or metastatic ER+ breast cancer. https://clinicaltrials.gov/study/NCT05080842?term=NCT05080842&rank=1.
ClinicalTrials.gov. A study of AC676 for the treatment of relapsed/refractory B-Cell malignancies. https://clinicaltrials.gov/study/NCT05780034?term=NCT05780034&rank=1.
ClinicalTrials.gov. A study of HSK29116 in adults with relapsed/refractory B-cell malignancies. https://clinicaltrials.gov/study/NCT04861779?term=NCT04861779&rank=1.
ClinicalTrials.gov. Study in subjects to evaluate the safety, tolerability and pharmacokinetics of GT20029. https://clinicaltrials.gov/study/NCT05428449?term=NCT05428449&rank=1.
ClinicalTrials.gov. A phase I study to assess the safety, pharmacokinetics, and anti-tumor activity of oral HP518 in mCRPC patients. https://clinicaltrials.gov/study/NCT06155084?term=NCT06155084&rank=1.
Acknowledgements
J.A.R.S. thanks the University of Coimbra for financial support. A.T.S.V. and S.P.S.P.M. acknowledge the Portuguese Science and Technology Foundation (FCT) for a doctoral scholarship 2023.03179.BD (https://doi.org/10.54499/2023.03179.BD) and SFRH/BD/138674/2018, respectively. The authors acknowledge Rita C. Guedes from the University of Lisboa (iMed.ULisboa) for creating the computational image of Fig. 2, using PyMOL software (Open-Source PyMOL is Copyright (C) Schrodinger, LLC). Graphical abstract, Figs. 1 and 3 were created in BioRender. Created in BioRender. Vicente, A. (2025) https://BioRender.com/y33i220. Created in BioRender. Vicente, A. (2025) https://BioRender.com/b19z226. Created in BioRender. Vicente, A. (2025) https://BioRender.com/f40h970.
Author information
Authors and Affiliations
Contributions
A.V. conceptualization, writing, editing and figures, S.M. conceptualization, figures and tables; J.S. conceptualization, review and supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vicente, A.T.S., Moura, S.P.S.P. & Salvador, J.A.R. Synthesis, biological evaluation and clinical trials of Cereblon-based PROTACs. Commun Chem 8, 218 (2025). https://doi.org/10.1038/s42004-025-01598-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-025-01598-9
